MolGAN:小分子图的隐式生成模型
摘要
图结构数据的深度生成模型为化学合成问题提供了一个新的角度:通过优化直接生成分子图的可微分模型,可以在离散且广阔的化学结构空间中避开昂贵的搜索过程。 我们引入了 MolGAN,这是一种用于小分子图的隐式、无似然生成模型,它避免了对昂贵的图匹配程序或先前基于似然的方法的节点排序启发式的需要。 我们的方法采用生成对抗网络(GAN)来直接对图结构数据进行操作。 我们将我们的方法与强化学习目标相结合,以鼓励生成具有特定所需化学性质的分子。 在 QM9 化学数据库的实验中,我们证明我们的模型能够生成接近 的有效化合物。 MolGAN 与最近使用基于字符串 (SMILES) 的分子表示的提议以及直接生成图形的基于可能性的方法相比,尽管容易受到模式崩溃的影响,但都具有优势。
1简介
对于从头药物设计(Schneider & Fechner,2005)等重要应用来说,寻找具有所需特性的新化合物是一项具有挑战性的任务。 可合成分子的空间是巨大的,在这个空间中的搜索被证明是非常困难的,这主要是由于其离散的性质。
深度生成模型开发的最新进展催生了一系列有前景的建议来解决这个问题。 该领域的大多数作品(Gómez-Bombarelli 等人,2016;Kusner 等人,2017;Guimaraes 等人,2017;Dai 等人,2018) 使用所谓的 SMILES 表示(Weininger, 1988) 分子:源自分子图的基于字符串的表示。 循环神经网络 (RNN) 是这些表示的理想候选者,因此,最近的工作遵循在此类编码上应用基于 RNN 的生成模型的方法。 然而,基于字符串的分子表示有一定的缺点:RNN 必须花费大量精力来学习表示的句法规则和顺序模糊性。 此外,这种方法不适用于通用(非分子)图。
SMILES 字符串是根据基于图形的分子表示生成的,因此在原始图形空间中工作具有消除额外开销的优点。 随着图训练深度学习领域的最新进展(Bronstein 等人,2017;Hamilton 等人,2017),直接基于图表示的深度生成模型成为一种可行的替代方案,并已在近期作品(Kipf & Welling, 2016; Johnson, 2017; Grover 等人, 2019; Li 等人, 2018b; Simonovsky & Komodakis, 2018; You 等人, 2018)。
基于似然的分子图生成方法(Li等人,2018b;Simonovsky & Komodakis,2018)然而,要么需要提供固定(或随机选择)的图有序表示,要么需要昂贵的图匹配评估生成分子的可能性的过程,因为对于小尺寸的图来说,所有可能的节点排序的评估已经是禁止的。
在这项工作中,我们通过利用隐式的、无似然的方法来回避这个问题,特别是生成对抗网络(GAN)(Goodfellow等人,2014),我们适应直接在图表示上工作。 我们进一步利用类似于 ORGAN (Guimaraes 等人,2017) 的强化学习(RL)目标来鼓励具有特定属性的分子的生成。
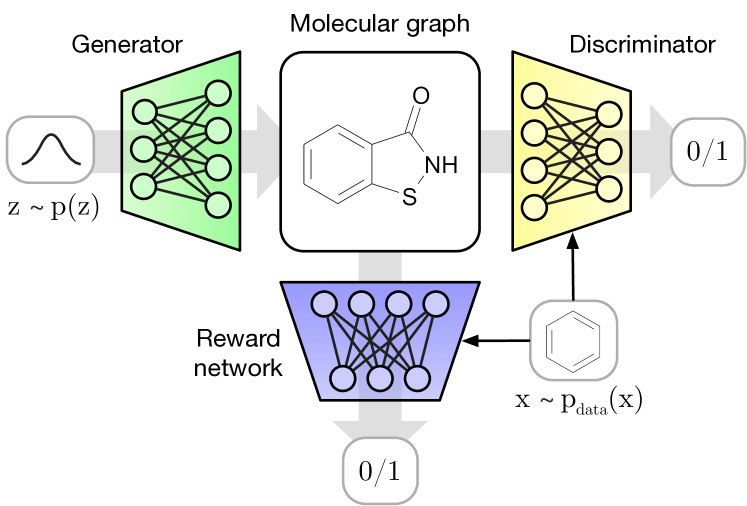
我们的分子 GAN (MolGAN) 模型(如图 1 所示)是第一个使用 GAN 在分子合成背景下生成图结构数据的模型(Goodfellow 等人,2014) 。 MolGAN 的生成模型可以一次性(即非顺序地)预测离散图结构,以提高计算效率,尽管顺序变体通常是可能的。 MolGAN 进一步利用基于图卷积层 的排列不变判别器和奖励网络(针对所需化学性质进行基于强化学习的优化)(Bruna 等人,2014;Duvenaud 等人,2015;Kipf & Welling,2017;Schlichtkrull等人,2017) 两者都直接在图结构表示上运行。
2 背景
2.1 分子作为图表
之前大多数分子数据深度生成模型(Gómez-Bombarelli 等人,2016;Kusner 等人,2017;Guimaraes 等人,2017;Dai 等人,2018) 诉诸于生成分子的 SMILES 表示。 然而,SMILES 语法对于微小的更改或错误并不稳健,这可能会导致生成无效或截然不同的结构。 语法 VAE (Kusner 等人, 2017) 通过限制生成过程遵循特定语法来缓解这个问题。
最近证明,直接在图空间中操作是分子数据生成建模的可行替代方案(Li等人,2018b;Simonovsky & Komodakis,2018),其额外好处是所有生成的输出是有效的图(但不一定是有效的分子)。
我们认为每个分子都可以用一个带有一组边和节点的无向图来表示。 每个原子对应一个节点,该节点与维一维一热向量关联,指示原子的类型。 我们进一步将每个原子键表示为与键类型 关联的边 。 对于具有 节点的分子图,我们可以将这种表示总结为节点特征矩阵 和邻接张量 ,其中 是一个 one-hot 向量,指示 和 之间的边的类型。
2.2 隐式方法与基于可能性的方法
基于似然的方法,例如变分自动编码器(VAE)(Kingma & Welling,2014;Rezende 等人,2014)通常比隐式生成模型(例如 GAN)更容易、更稳定的优化(Goodfellow 等人,2014)。 然而,在生成图结构数据时,我们希望对图的(有序)矩阵表示中的节点重新排序保持不变,这要求我们执行极其昂贵的图匹配过程 (Simonovsky & Komodakis,2018 ) 或明确评估所有可能的节点排列的可能性。
通过诉诸隐式生成模型,特别是 GAN 框架,我们规避了对显式可能性的需求。 而通过利用图卷积(Bruna等人,2014;Duvenaud等人,2015;Kipf&Welling,2017)和节点聚合算子(Li 等人, 2016),生成器在生成图时仍然需要决定特定的节点顺序。 然而,由于我们不提供可能性,生成器可以自由地为手头的任务选择任何合适的顺序。 下面我们对 GAN 进行简要介绍。
生成对抗网络
GAN (Goodfellow 等人,2014) 是隐式生成模型,因为它们允许推断模型参数,而不需要指定可能性。
GAN 由两个主要组件组成:生成模型 ,它从数据分布之前学习映射以采样新数据点;以及判别模型 ,它学习分类样本是否来自数据分布而不是来自 。 这两个模型以神经网络的形式实现,并通过随机梯度下降 (SGD) 同时进行训练。 和有不同的目标,他们可以被视为极小极大游戏中的两个玩家
| (1) |
其中 尝试生成样本来欺骗鉴别器,而 尝试正确区分样本。 为了防止模式崩溃等不良行为(Salimans等人,2016)并稳定学习,我们使用小批量判别(Salimans等人,2016)和改进的WGAN(Gulrajani 等人, 2017),一种替代且更稳定的 GAN 模型,可最大限度地减少更适合的分歧。
改进的WGAN
WGAN (Arjovsky 等人,2017) 最小化两个概率分布之间定义的 Earth Mover (EM) 距离(也称为 Wasserstein-1 距离)的近似值。 形式上,使用 Kantorovich-Rubinstein 对偶性的 和 之间的 Wasserstein 距离为
| (2) |
其中,对于 WGAN, 是经验分布, 是生成器分布。 请注意,上界是针对某些 的所有 K-Lipschitz 函数。
Gulrajani 等人 (2017) 引入梯度惩罚作为 1-Lipschitz 连续性的替代软约束,作为对原始 WGAN 的梯度裁剪方案的改进。 关于生成器的损失与 WGAN 中的相同,但是关于鉴别器的损失函数修改为
| (3) |
其中, 是一个超参数(我们和原论文一样使用 ), 是 和 与 之间的采样线性组合,因此 与 之间的采样线性组合。
2.3确定性策略梯度
GAN 生成器学习从先验分布到数据分布的转换。 因此,生成的样本类似于数据样本。 然而,在从头药物设计方法中,我们不仅对生成化学上有效的化合物感兴趣,而且希望它们具有一些有用的特性(例如,易于合成)。 因此,我们还使用强化学习针对一些不可微指标优化生成过程。
在强化学习中,随机策略由 表示,它是 中的参数概率分布,根据状态 选择分类行动 。相反,确定性策略由 表示,它确定性地输出一个操作。
在最初的实验中,我们探索了将 REINFORCE (Williams,1992) 与随机策略结合使用,该随机策略将图生成建模为一组分类选择(动作)。 然而,我们发现,由于一次性生成图时动作空间高维,它的收敛性很差。 相反,我们的方法基于确定性策略梯度算法,众所周知,该算法在高维动作空间中表现良好(Silver 等人,2014)。 特别是,我们采用了 Lillicrap 等人 (2016) 引入的深度确定性策略梯度 (DDPG) 的简化版本,这是一种非策略行为者批评算法,它使用确定性策略梯度来最大化预期的未来回报。
在我们的例子中,策略是 GAN 生成器 ,它采用先验样本 作为输入,而不是环境状态 ,并输出作为动作的分子图 ()。 此外,我们不对情节进行建模,因此无需评估状态-动作组合的质量,因为它仅取决于图 。 因此,我们引入了奖励函数 的可学习且可微的近似值来预测即时奖励,并且我们通过基于外部系统提供的真实奖励的均方误差目标来训练它(例如,分子的可合成性分数)。 然后,我们通过 训练生成器最大化预测奖励,它是可微分的,为策略提供了朝向所需指标的梯度。
3型号
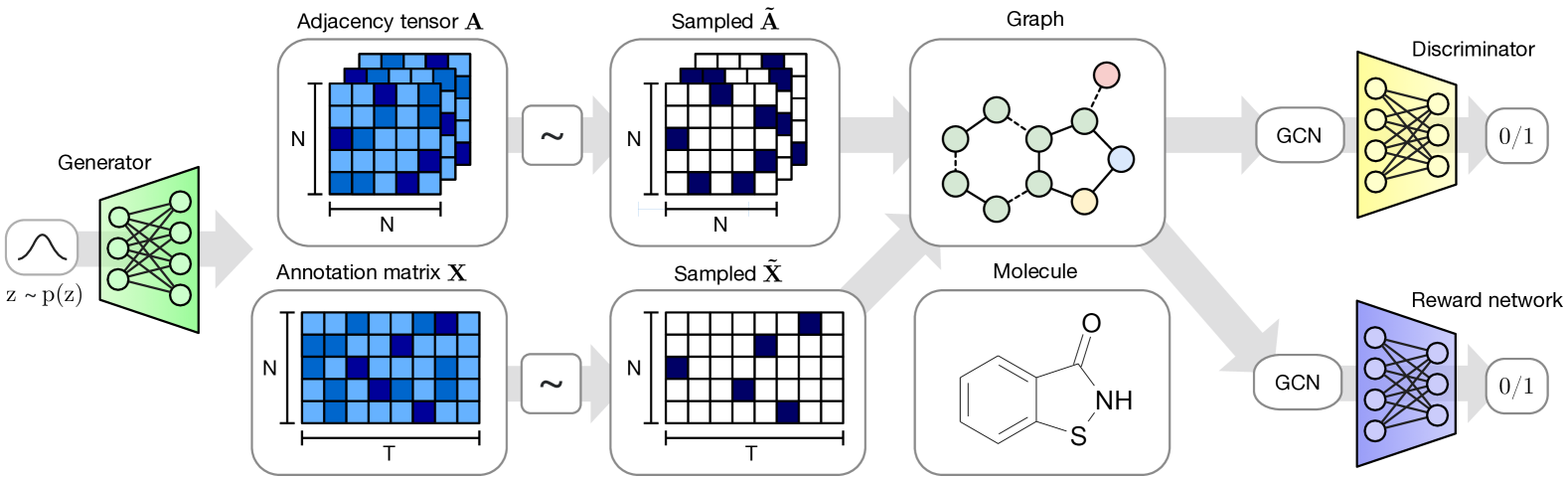
MolGAN 架构(图2)由三个主要组件组成:生成器、鉴别器和奖励网络 。
生成器从先验分布中获取样本并生成表示分子的带注释的图 。 的节点和边分别与表示原子类型和键类型的注释相关联。 鉴别器从数据集和生成器中获取样本,并学习区分它们。 和 均使用改进的 WGAN 进行训练,以便生成器学习匹配经验分布并最终输出有效分子。
奖励网络用于近似样本的奖励函数,并使用强化学习针对不可微指标优化分子生成。 数据集和生成的样本是 的输入,但是与判别器不同,它为它们分配分数(例如,生成的分子溶于水的可能性有多大)。 奖励网络学习为每个分子分配奖励,以匹配外部软件提供的分数111我们使用 RDKit 开源化学信息学软件:http://www.rdkit.org。. 请注意,当 MolGAN 输出无效分子时,无法分配奖励,因为该图甚至不是化合物。 因此,对于无效的分子图,我们分配零奖励。
鉴别器使用 WGAN 目标进行训练,而生成器使用 WGAN 损失和 RL 损失的线性组合:
| (4) |
其中 是一个超参数,用于调节两个组件之间的权衡。
3.1生成器
获取从标准正态分布 采样的 D 维向量 并输出图形。 虽然最近的工作表明使用基于 RNN 的生成模型生成小尺寸图是可行的(Johnson,2017;You 等人,2018;Li 等人,2018a,b)为简单起见,使用生成模型,使用简单的多层感知器 (MLP) 一次性预测整个图,与 Simonovsky & Komodakis (2018) 中的做法类似。 虽然这将我们的研究限制在预先选择的最大尺寸的图表上,但我们发现它的优化速度明显更快且更容易。
我们将域限制为有限数量节点的图,并且对于每个 , 输出两个连续且密集的对象: 定义原子类型和 定义债券类型(请参阅第 2.1 节)。 和 都有概率解释,因为每个节点和边类型都用类型上的分类分布的概率表示。 为了生成分子,我们分别通过 和 的分类采样获得离散、稀疏的对象 和 。 我们重载符号,并使用二进制 和 表示数据集中的样本。
由于这种离散化过程是无差别的,我们探索了三种模型变化,以实现基于梯度的训练:我们可以 i) 在向前传递过程中直接使用连续对象 和 (即 和 ),ii) 在将 和 传递给 和 之前给它们添加 Gumbel 噪声,以便使生成随机,同时仍然传递连续对象(即 和 或 iii)、和),或 iii) 使用基于分类重参数化的 Gumbel-Softmax (Jang 等人,2017 年;Maddison 等人,2017 年)的直通梯度、2017),即我们在前向传递中使用分类分布的样本形式(即 和 ),在后向传递中使用连续的松弛值(即原始的 和 )。
3.2 判别器和奖励网络
判别器 和奖励网络 都接收一个图作为输入,并各自输出一个标量值。 我们为两个网络选择相同的架构,但它们之间不共享参数。 一系列图卷积层使用图邻接张量对节点信号进行卷积。 我们的模型基于 Relational-GCN (Schlichtkrull 等人,2017),这是一种支持多种边缘类型的图卷积网络。 在每一层,节点的特征表示根据以下方式进行卷积/传播:
| (5) |
其中是层节点的信号,是充当自连接的线性变换函数各层之间。 我们进一步为每一层使用边缘类型特定的仿射函数。 表示节点 的邻居集合。 归一化因子 确保激活处于相似的规模,而与邻居的数量无关。
经过图卷积的几层传播后,按照Li等人(2016),我们将节点嵌入聚合到图级表示向量中:
| (6) |
其中 是逻辑 sigmoid 函数, 和 是具有线性输出层的 MLP, 表示逐元素乘法。 然后, 是图 的向量表示,并由 MLP 进一步处理,为鉴别器生成图级标量输出 和 奖励网络。
4相关工作
Guimaraes 等人 (2017) 的目标强化生成对抗网络 (ORGAN) 是与我们最接近的相关工作。 他们的模型依靠 SeqGAN (Yu 等人,2017) 来对抗性地学习输出序列,同时使用 REINFORCE (Williams,1992) 优化化学指标。 与我们的方法的主要区别在于,它们将 SMILES 序列建模为分子表示而不是图形,并且它们的 RL 组件使用 REINFORCE,而我们使用 DDPG。 Segler 等人 (2018) 还通过使用蒙特卡罗树搜索 (MCTS) 结合扩展策略网络搜索逆合成路线,将强化学习用于药物发现。
其他几项工作也探索了分子 SMILES 表示的训练生成模型:CharacterVAE (Gómez-Bombarelli 等人, 2016) 是第一个基于具有循环编码器和解码器网络的 VAE 的模型。 GrammarVAE (Kusner 等人, 2017) 和 SDVAE (Dai 等人, 2018) 约束解码过程遵循特定的句法和语义规则。
相关的研究方向考虑训练深度生成模型来直接输出图结构数据。 一些工作探索了利用图卷积进行图内链接预测的自动编码器架构(Kipf & Welling,2016;Grover 等人,2019;Davidson 等人,2018)。 约翰逊(2017);李等人 (2018b);你等人 (2018);另一方面,Li 等人 (2018a) 开发了基于似然的方法,以顺序方式直接输出任意大小的图。 一些相关的工作已经探索了扩展 VAE 来直接生成图,例子包括 GraphVAE (Simonovsky & Komodakis, 2018)、Junction Tree VAE (Jin 等人, 2018) 和NeVAE (Samanta 等人, 2018) 模型。
对于图中的链接预测,文献中引入了一系列对抗方法(Minervini 等人,2017;Wang 等人,2018;Bojchevski 等人,2018)。 然而,此类模型不适合从头开始生成分子图,这使得直接比较不可行。
5实验
我们使用 QM9 (Ramakrishnan 等人,2014) 化学数据库在既定基准上进行了一系列实验,将 MolGAN 与最近基于神经网络的药物生成模型进行了比较。 我们首先重点研究 参数的影响,以找到 GAN 和 RL 目标之间的最佳权衡(参见第 5.1 节)。 然后,我们将 MolGAN 与 ORGAN (Guimaraes 等人,2017) 进行比较,因为它是与我们最相关的工作:ORGAN 是一个在 SMILES 表示上运行的顺序生成模型,通过 RL 目标针对多种化学性质进行优化(参见第 5.2 节)。 我们还将我们的模型与变分自动编码方法(第5.3节)进行比较,例如CharacterVAE (Gómez-Bombarelli等人,2016),GrammarVAE (Kusner等人,2017) ),以及最近的基于图的生成模型:GraphVAE (Simonovsky & Komodakis,2018)。
数据集
在所有实验中,我们使用 QM9 (Ramakrishnan 等人,2014),这是 GDB-17 化学数据库 1664 亿分子的一个子集(Ruddigkeit 等人,2012)。 QM9 包含 133,885 种有机化合物,最多 9 个重原子:碳 (C)、氧 (O)、氮 (N) 和氟 (F)。
生成器架构
所有实验的生成器架构都是固定的。 我们使用 作为节点的最大数量, 作为原子类型的数量(C、O、N、F 和一个填充符号), 作为键类型的数量(单键、双键、三键和无键)。 这些维度足以覆盖 QM9 中的所有分子。 生成器采用从标准正态分布 采样的 32 维向量,并分别使用 个隐藏单元的 3 层 MLP 对其进行处理,其中 作为激活函数。 最终,最后一层被线性投影以匹配 和 维度,并通过 操作对其最后一个维度进行归一化 ( )。
判别器和奖励网络架构
评价措施
我们衡量 Samanta 等人 (2018) 中定义的以下统计数据:有效性、新颖性和独特性。 有效性定义为有效分子数与所有生成分子数之间的比率。 新颖性衡量数据集中不存在的有效样本集与有效样本总数之间的比率。 最后,唯一性定义为唯一样本数与有效样本数之间的比率,它衡量抽样过程中的变异程度。
训练
在所有实验中,我们使用的批量大小为 32,并使用 Adam (Kingma & Ba, 2015) 优化器进行训练,学习率为 。 对于每个设置,我们对辍学率 (Srivastava等人,2014)以及离散化变化(如第3.1 我们总是根据我们正在优化的内容来报告最佳模型的结果(例如,在优化溶解度时,我们报告具有最高溶解度分数的模型 - 当没有优化指标时,我们报告具有最高个体分数总和的模型)。 尽管使用 WGAN 在某种程度上应该可以防止诸如模式崩溃(Salimans等人,2016)之类的不良行为,但我们注意到我们的模型遇到了这个问题。 我们将在未来的工作中解决这个问题。 作为一个简单的对策,我们采用提前停止的方式,每 10 个 epoch 进行评估,以避免完全崩溃的模式。 特别是,我们使用独特的分数来衡量模型的崩溃程度,因为它本质上表明了生成过程中存在多少多样性。 我们设置了 2% 的任意阈值,低于该阈值我们认为模型崩溃并停止训练。
在我们工作的早期阶段,我们注意到奖励网络需要几个时期的预训练才能用于将梯度传播到生成器,否则生成器很容易发散。 我们认为发生这种情况是因为在训练开始时,没有准确地预测奖励,然后它没有很好地优化生成器。 因此,在每个实验中,我们在没有 RL 组件的情况下训练前半个 epoch 的生成器,但仅使用 WGAN 目标。 我们在这些时期训练奖励网络,但没有使用强化学习损失来训练生成器。 对于后半个时期,我们使用方程 4 中的组合损失。
5.1 的效果
正如 Guimaraes 等人 (2017) 中一样, 超参数控制最大化期望目标和调节生成器以匹配数据分布之间的权衡。 我们研究了 对溶解度度量的影响(更多详细信息,请参阅第 5.2 节)。 我们在 Guimaraes 等人 (2017) 中使用的 QM9 的 5k 子集上训练了 300 个 epoch(其中 150 个用于预训练)。 我们在所有其他实验(第 5.2 节和第 5.3 节)中使用最佳 参数,该参数是通过具有最大有效、唯一、新颖和可溶性得分之和的模型确定的,无需进行任何进一步搜索。
结果
我们在表 1 中报告结果。 我们观察到 值越低,有效性得分越高的明显趋势。 这可能是由于有效分子的隐式优化,因为无效分子在训练期间获得零奖励。 因此,如果 RL 损失分量很强,则生成器会被优化以生成大部分有效的分子图。 相反,似乎并不主要影响独特和新颖的乐谱。 请注意,这些分数并未直接或间接优化,因此它们是模型架构、超参数和训练过程的结果。 事实上,独特的分数总是接近 2%(这是我们的阈值),表明如果我们不应用提前停止,模型就会崩溃(即使在仅 RL 的情况下)。
我们还运行 而无需从预训练模型开始。 我们观察到它成功地针对所需的指标进行了优化,但它崩溃了,输出的样本非常少(即,唯一得分低)。 这种行为可能表明预训练是使用 RL 之前匹配数据分布的基础,因为 GAN 的作用是针对多样性进行正则化。
由于 控制 WGAN 和 RL 损失之间的权衡,因此 (即训练后半部分仅使用 RL)产生最高的有效和最高值也就不足为奇了。与其他值相比的溶解度分数。 分数总和最高的 值为 。 我们使用这个值进行后续实验。
| Algorithm | Valid | Unique | Novel | Sol. |
|---|---|---|---|---|
| (full RL)* | 100.0 | 0.03 | 100.0 | 0.98 |
| (full RL) | 99.8 | 2.3 | 97.9 | 0.86 |
| 98.2 | 2.2 | 98.1 | 0.74 | |
| 92.2 | 2.7 | 95.0 | 0.67 | |
| 87.3 | 3.2 | 87.2 | 0.56 | |
| 86.6 | 2.1 | 87.5 | 0.48 | |
| (no RL) | 87.7 | 2.9 | 97.7 | 0.54 |
5.2目标优化
| Objective | Algorithm | Valid (%) | Unique (%) | Time (h) | Diversity | Druglikeliness | Synthesizability | Solubility |
|---|---|---|---|---|---|---|---|---|
| Druglikeliness | ORGAN | 88.2 | 69.4* | 9.63* | 0.55 | 0.52 | 0.32 | 0.35 |
| OR(W)GAN | 85.0 | 8.2* | 10.06* | 0.95 | 0.60 | 0.54 | 0.47 | |
| Naive RL | 97.1 | 54.0* | 9.39* | 0.80 | 0.57 | 0.53 | 0.50 | |
| MolGAN | 99.9 | 2.0 | 1.66 | 0.95 | 0.61 | 0.68 | 0.52 | |
| MolGAN (QM9) | 100.0 | 2.2 | 4.12 | 0.97 | 0.62 | 0.59 | 0.53 | |
| Synthesizability | ORGAN | 96.5 | 45.9* | 8.66* | 0.92 | 0.51 | 0.83 | 0.45 |
| OR(W)GAN | 97.6 | 30.7* | 9.60* | 1.00 | 0.20 | 0.75 | 0.84 | |
| Naive RL | 97.7 | 13.6* | 10.60* | 0.96 | 0.52 | 0.83 | 0.46 | |
| MolGAN | 99.4 | 2.1 | 1.04 | 0.75 | 0.52 | 0.90 | 0.67 | |
| MolGAN (QM9) | 100.0 | 2.1 | 2.49 | 0.95 | 0.53 | 0.95 | 0.68 | |
| Solubility | ORGAN | 94.7 | 54.3* | 8.65* | 0.76 | 0.50 | 0.63 | 0.55 |
| OR(W)GAN | 94.1 | 20.8* | 9.21* | 0.90 | 0.42 | 0.66 | 0.54 | |
| Naive RL | 92.7 | 100.0* | 10.51* | 0.75 | 0.49 | 0.70 | 0.78 | |
| MolGAN | 99.8 | 2.3 | 0.58 | 0.97 | 0.45 | 0.42 | 0.86 | |
| MolGAN (QM9) | 99.8 | 2.0 | 1.62 | 0.99 | 0.44 | 0.22 | 0.89 | |
| All/Alternated | ORGAN | 96.1 | 97.2* | 10.2* | 0.92 | 0.52 | 0.71 | 0.53 |
| All/Simultaneously | MolGAN | 97.4 | 2.4 | 2.12 | 0.91 | 0.47 | 0.84 | 0.65 |
| All/Simultaneously | MolGAN (QM9) | 98.0 | 2.3 | 5.83 | 0.93 | 0.51 | 0.82 | 0.69 |
与之前的实验类似,我们在 5k QM9 子集上训练模型 300 个周期,同时优化与 Guimaraes 等人 (2017) 相同的目标,以与他们的工作进行比较。 此外,我们还报告了经过 30 个 epoch 训练的完整数据集的结果(请注意,完整数据集比子集大 20 倍)。 所有分数均标准化为 范围内。 我们为无效化合物分配零分(即,隐含地我们也在优化有效性分数)。 我们选择优化以下目标,这些目标代表了药物发现通常所需的品质:
药物相似性:
化合物成为药物的可能性有多大。 药物相似性定量估计 (QED) 评分通过合意性评分的加权几何平均值来量化化合物质量,捕获多种药物特性的基础数据分布(Bickerton 等人,2012)。
溶解度:
分子的亲水程度。 对数辛醇-水分配系数 (logP) 定义为溶质的两种溶剂之间的浓度比的对数 (Comer & Tam, 2001)。
可合成性:
该指标量化了分子合成的难易程度。 综合可访问性分数(Ertl & Schuffenhauer,2009)是一种以概率方式估计综合难易程度的方法。
我们还测量了多样性分数,但没有对其进行优化,该分数表明分子相对于数据集具有多样性的可能性有多大。 该度量比较样本之间的子结构和数据集中的随机子集,指示有多少重复。
为了进行评估,我们报告了 6400 个采样化合物的平均分数,如(Guimaraes 等人,2017)。 此外,我们重新运行(Guimaraes等人,2017)的实验来计算独特的分数和执行时间,因为它没有报告。 与 ORGAN 不同的是,为了优化所有目标,我们不会在训练期间单独优化它们,这在我们的例子中是不可能的,因为奖励网络特定于单一类型的奖励。 因此,我们改为优化联合奖励,将其定义为所有目标的乘积(位于 内)。
结果
结果报告于表2中。 附录中提供了定性样本(图3)。 我们观察到,MolGAN 模型在训练结束时总是收敛到非常高的有效性输出 。 正如在之前的实验中观察到的那样,这是一致的,因为这里也存在隐式的有效性优化。 此外,在所有单一指标设置中,我们的模型在有效分数以及我们优化的所有三个目标分数方面都击败了 ORGAN 模型。
我们认为这主要是由于两个因素:i)直观上来说,优化预测为单个样本的分子图应该比优化输出字符序列的 RNN 模型更容易,ii)使用确定性策略梯度而不是 REINFORCE 有效地提供了更好的梯度,它改进了指标的采样过程,同时惩罚无效图。
在完整的 QM9 数据集上进行训练,训练次数减少了 10 倍,进一步提高了几乎所有分数的结果。 在训练过程中,我们的算法观察更多不同的样本,因此它可以用更少的迭代次数很好地学习。 此外,它可以观察具有更多样化结构和性质的分子。
正如之前在 5.1 节中观察到的,在这个实验中,唯一得分始终接近 2%,这证实了我们的假设,即我们的模型容易受到模式崩溃的影响。 ORGAN 基线的情况并非如此。 在采样过程中,ORGAN 生成最多 51 个字符的序列,这使其能够生成更大的分子,而我们的模型(根据选择)被限制为最多生成 9 个原子。 这解释了独特分数的差异,因为在较小的空间中生成不同分子的机会要低得多。 请注意,在 ORGAN 中,强化学习组件依赖于 REINFORCE,并且独特的分数经过优化,惩罚非独特的输出,而我们没有这样做。
就训练时间而言,在 5k 数据集上训练时,我们的模型大幅优于 ORGAN(每种设置至少快 5 倍),因为我们不依赖顺序生成或区分。 ORGAN 和 MolGAN 的参数数量相当,后者大约更大 。
5.3 VAE 基线
| Algorithm | Valid | Unique | Novel |
|---|---|---|---|
| CharacterVAE | 10.3 | 67.5 | 90.0 |
| GrammarVAE | 60.2 | 9.3 | 80.9 |
| GraphVAE | 55.7 | 76.0 | 61.6 |
| GraphVAE/imp | 56.2 | 42.0 | 75.8 |
| GraphVAE NoGM | 81.0 | 24.1 | 61.0 |
| MolGAN | 98.1 | 10.4 | 94.2 |
在本实验中,我们将 MolGAN 与最近利用 VAE 的基于可能性的方法进行比较。 我们报告了与 CharacterVAE (Gómez-Bombarelli 等人, 2016)、GrammarVAE (Kusner 等人, 2017) 和 GraphVAE (Simonovsky & Komodakis, 2018) 的比较)。 在这里,我们使用完整的 QM9 数据集进行训练。 当然,我们只与衡量生成过程质量的指标进行比较,因为可能性不是在 MolGAN 中直接计算的。 此外,除了有效性之外,我们不会优化任何特定的化学性质(即,我们不会优化上述任何指标,但我们会针对化学上有效的化合物进行优化)。 最终评估分数是个随机样本的平均值。 样本数量与之前的实验有所不同,符合Simonovsky & Komodakis (2018)中的设置。
结果
尽管 MolGAN 的独特得分比 GrammarVAE 略高,但其他基线在该得分方面更胜一筹。 尽管在这里我们不认为我们的模型崩溃了,但如此低的分数证实了我们的假设,即我们的模型容易出现模式崩溃。 另一方面,与基于 VAE 的基线相比,我们观察到明显更高的有效性分数。为了验证采样的独特分子(大部分)是新颖的,而不是简单地从数据集中记住的,我们还测量了有多少独特 分子对于我们的模型来说也是新颖的。 该分数为 97%,表明几乎所有独特分子确实都是新颖的,而 MolGAN 不会遇到此类问题。
与我们的方法不同,VAE 优化了证据下限 (ELBO),并且没有显式或隐式输出有效性的优化。 此外,由于 ELBO 的一部分最大化了观测值的重建,因此采样过程的新颖性预计不会很高,因为它没有经过优化。 然而,在所有报告的方法中,新颖性是,对于CharacterVAE,新颖性是。 尽管CharacterVAE可以获得很高的新颖性分数,但它在有效性方面表现不佳。 另一方面,MolGAN 获得了较高的有效性和新颖性分数。
6 结论
在这项工作中,我们引入了 MolGAN:一种用于小尺寸分子图的隐式生成模型。 通过 GAN 和 RL 目标的联合训练,我们的模型能够生成比之前基于 VAE 的生成模型具有更高有效性和新颖性的分子图,同时不需要依赖于排列的似然函数。 与最近用于分子生成的基于 SMILES 的序列 GAN 模型相比,MolGAN 可以获得更高的化学属性分数(例如溶解度),同时允许至少 5 倍的训练时间。
我们目前的 MolGAN 表述的一个核心限制是它们对模式崩溃的敏感性:GAN 和 RL 目标都不鼓励生成多样化和非唯一的输出,因此模型往往会被拉向仅涉及很少样本变异性的解决方案。 如果不尽早停止训练,最终只会产生少数不同的分子。
我们认为这个问题可以在未来的工作中得到解决,例如通过仔细设计奖励函数或某种形式的预训练。 MolGAN 框架与已建立的化学合成基准数据集相结合,为改进 GAN 在模式崩溃问题方面的稳定性提供了一个新的测试平台。 我们相信,即使在生成分子图的范围之外,从此类评估中获得的见解也将对社区有价值。 最后,我们有望在 MolGAN 框架内探索替代的生成架构,例如基于循环图的生成模型(Johnson,2017;Li 等人,2018b;You 等人,2018),如我们当前对邻接张量的一次性预测很可能仅适用于小尺寸的图。
致谢
作者要感谢 Luca Falorsi、Tim R. Davidson、Herke van Hoof 和 Max Welling 的有益讨论和反馈。 T.K. 由 SAP SE Berlin 提供支持。
参考
- Arjovsky et al. (2017) Arjovsky, Martín, Chintala, Soumith, and Bottou, Léon. Wasserstein generative adversarial networks. In Precup, Doina and Teh, Yee Whye (eds.), Proceedings of the 34th International Conference on Machine Learning, ICML 2017, Sydney, NSW, Australia, 6-11 August 2017, volume 70 of Proceedings of Machine Learning Research, pp. 214–223. PMLR, 2017. URL http://proceedings.mlr.press/v70/arjovsky17a.html.
- Bickerton et al. (2012) Bickerton, G Richard, Paolini, Gaia V, Besnard, Jérémy, Muresan, Sorel, and Hopkins, Andrew L. Quantifying the chemical beauty of drugs. Nature chemistry, 4(2):90, 2012.
- Bojchevski et al. (2018) Bojchevski, Aleksandar, Shchur, Oleksandr, Zügner, Daniel, and Günnemann, Stephan. Netgan: Generating graphs via random walks. In Dy, Jennifer G. and Krause, Andreas (eds.), Proceedings of the 35th International Conference on Machine Learning, ICML 2018, Stockholmsmässan, Stockholm, Sweden, July 10-15, 2018, volume 80 of Proceedings of Machine Learning Research, pp. 609–618. PMLR, 2018. URL http://proceedings.mlr.press/v80/bojchevski18a.html.
- Bronstein et al. (2017) Bronstein, Michael M, Bruna, Joan, LeCun, Yann, Szlam, Arthur, and Vandergheynst, Pierre. Geometric deep learning: going beyond euclidean data. IEEE Signal Processing Magazine, 34(4):18–42, 2017.
- Bruna et al. (2014) Bruna, Joan, Zaremba, Wojciech, Szlam, Arthur, and LeCun, Yann. Spectral networks and locally connected networks on graphs. In Bengio, Yoshua and LeCun, Yann (eds.), 2nd International Conference on Learning Representations, ICLR 2014, Banff, AB, Canada, April 14-16, 2014, Conference Track Proceedings, 2014. URL http://arxiv.org/abs/1312.6203.
- Comer & Tam (2001) Comer, John and Tam, Kin. Lipophilicity profiles: theory and measurement. Wiley-VCH: Zürich, Switzerland, 2001.
- Dai et al. (2018) Dai, Hanjun, Tian, Yingtao, Dai, Bo, Skiena, Steven, and Song, Le. Syntax-directed variational autoencoder for molecule generation. In International Conference on Machine Learning, 2018.
- Davidson et al. (2018) Davidson, Tim R., Falorsi, Luca, Cao, Nicola De, Kipf, Thomas, and Tomczak, Jakub M. Hyperspherical variational auto-encoders. In Globerson, Amir and Silva, Ricardo (eds.), Proceedings of the Thirty-Fourth Conference on Uncertainty in Artificial Intelligence, UAI 2018, Monterey, California, USA, August 6-10, 2018, pp. 856–865. AUAI Press, 2018. URL http://auai.org/uai2018/proceedings/papers/309.pdf.
- Duvenaud et al. (2015) Duvenaud, David, Maclaurin, Dougal, Aguilera-Iparraguirre, Jorge, Gómez-Bombarelli, Rafael, Hirzel, Timothy, Aspuru-Guzik, Alán, and Adams, Ryan P. Convolutional networks on graphs for learning molecular fingerprints. In Cortes, Corinna, Lawrence, Neil D., Lee, Daniel D., Sugiyama, Masashi, and Garnett, Roman (eds.), Advances in Neural Information Processing Systems 28: Annual Conference on Neural Information Processing Systems 2015, December 7-12, 2015, Montreal, Quebec, Canada, pp. 2224–2232, 2015. URL https://proceedings.neurips.cc/paper/2015/hash/f9be311e65d81a9ad8150a60844bb94c-Abstract.html.
- Ertl & Schuffenhauer (2009) Ertl, Peter and Schuffenhauer, Ansgar. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. Journal of cheminformatics, 1(1):8, 2009.
- Gómez-Bombarelli et al. (2016) Gómez-Bombarelli, Rafael, Wei, Jennifer N, Duvenaud, David, Hernández-Lobato, José Miguel, Sánchez-Lengeling, Benjamín, Sheberla, Dennis, Aguilera-Iparraguirre, Jorge, Hirzel, Timothy D, Adams, Ryan P, and Aspuru-Guzik, Alán. Automatic chemical design using a data-driven continuous representation of molecules. ACS Central Science, 2016.
- Goodfellow et al. (2014) Goodfellow, Ian J., Pouget-Abadie, Jean, Mirza, Mehdi, Xu, Bing, Warde-Farley, David, Ozair, Sherjil, Courville, Aaron C., and Bengio, Yoshua. Generative adversarial nets. In Ghahramani, Zoubin, Welling, Max, Cortes, Corinna, Lawrence, Neil D., and Weinberger, Kilian Q. (eds.), Advances in Neural Information Processing Systems 27: Annual Conference on Neural Information Processing Systems 2014, December 8-13 2014, Montreal, Quebec, Canada, pp. 2672–2680, 2014. URL https://proceedings.neurips.cc/paper/2014/hash/5ca3e9b122f61f8f06494c97b1afccf3-Abstract.html.
- Grover et al. (2019) Grover, Aditya, Zweig, Aaron, and Ermon, Stefano. Graphite: Iterative generative modeling of graphs. In Chaudhuri, Kamalika and Salakhutdinov, Ruslan (eds.), Proceedings of the 36th International Conference on Machine Learning, ICML 2019, 9-15 June 2019, Long Beach, California, USA, volume 97 of Proceedings of Machine Learning Research, pp. 2434–2444. PMLR, 2019. URL http://proceedings.mlr.press/v97/grover19a.html.
- Guimaraes et al. (2017) Guimaraes, Gabriel Lima, Sanchez-Lengeling, Benjamin, Farias, Pedro Luis Cunha, and Aspuru-Guzik, Alán. Objective-reinforced generative adversarial networks (organ) for sequence generation models. ArXiv preprint, abs/1705.10843, 2017. URL https://arxiv.org/abs/1705.10843.
- Gulrajani et al. (2017) Gulrajani, Ishaan, Ahmed, Faruk, Arjovsky, Martín, Dumoulin, Vincent, and Courville, Aaron C. Improved training of wasserstein gans. In Guyon, Isabelle, von Luxburg, Ulrike, Bengio, Samy, Wallach, Hanna M., Fergus, Rob, Vishwanathan, S. V. N., and Garnett, Roman (eds.), Advances in Neural Information Processing Systems 30: Annual Conference on Neural Information Processing Systems 2017, December 4-9, 2017, Long Beach, CA, USA, pp. 5767–5777, 2017. URL https://proceedings.neurips.cc/paper/2017/hash/892c3b1c6dccd52936e27cbd0ff683d6-Abstract.html.
- Hamilton et al. (2017) Hamilton, William L, Ying, Rex, and Leskovec, Jure. Representation learning on graphs: Methods and applications. ArXiv preprint, abs/1709.05584, 2017. URL https://arxiv.org/abs/1709.05584.
- Jang et al. (2017) Jang, Eric, Gu, Shixiang, and Poole, Ben. Categorical reparameterization with gumbel-softmax. In 5th International Conference on Learning Representations, ICLR 2017, Toulon, France, April 24-26, 2017, Conference Track Proceedings. OpenReview.net, 2017. URL https://openreview.net/forum?id=rkE3y85ee.
- Jin et al. (2018) Jin, Wengong, Barzilay, Regina, and Jaakkola, Tommi S. Junction tree variational autoencoder for molecular graph generation. In Dy, Jennifer G. and Krause, Andreas (eds.), Proceedings of the 35th International Conference on Machine Learning, ICML 2018, Stockholmsmässan, Stockholm, Sweden, July 10-15, 2018, volume 80 of Proceedings of Machine Learning Research, pp. 2328–2337. PMLR, 2018. URL http://proceedings.mlr.press/v80/jin18a.html.
- Johnson (2017) Johnson, Daniel D. Learning graphical state transitions. In 5th International Conference on Learning Representations, ICLR 2017, Toulon, France, April 24-26, 2017, Conference Track Proceedings. OpenReview.net, 2017. URL https://openreview.net/forum?id=HJ0NvFzxl.
- Kingma & Ba (2015) Kingma, Diederik P. and Ba, Jimmy. Adam: A method for stochastic optimization. In Bengio, Yoshua and LeCun, Yann (eds.), 3rd International Conference on Learning Representations, ICLR 2015, San Diego, CA, USA, May 7-9, 2015, Conference Track Proceedings, 2015. URL http://arxiv.org/abs/1412.6980.
- Kingma & Welling (2014) Kingma, Diederik P. and Welling, Max. Auto-encoding variational bayes. In Bengio, Yoshua and LeCun, Yann (eds.), 2nd International Conference on Learning Representations, ICLR 2014, Banff, AB, Canada, April 14-16, 2014, Conference Track Proceedings, 2014. URL http://arxiv.org/abs/1312.6114.
- Kipf & Welling (2016) Kipf, Thomas N and Welling, Max. Variational graph auto-encoders. In NIPS Bayesian Deep Learning Workshop, 2016.
- Kipf & Welling (2017) Kipf, Thomas N. and Welling, Max. Semi-supervised classification with graph convolutional networks. In 5th International Conference on Learning Representations, ICLR 2017, Toulon, France, April 24-26, 2017, Conference Track Proceedings. OpenReview.net, 2017. URL https://openreview.net/forum?id=SJU4ayYgl.
- Kusner et al. (2017) Kusner, Matt J., Paige, Brooks, and Hernández-Lobato, José Miguel. Grammar variational autoencoder. In Precup, Doina and Teh, Yee Whye (eds.), Proceedings of the 34th International Conference on Machine Learning, ICML 2017, Sydney, NSW, Australia, 6-11 August 2017, volume 70 of Proceedings of Machine Learning Research, pp. 1945–1954. PMLR, 2017. URL http://proceedings.mlr.press/v70/kusner17a.html.
- Li et al. (2018a) Li, Yibo, Zhang, Liangren, and Liu, Zhenming. Multi-objective de novo drug design with conditional graph generative model. ArXiv preprint, abs/1801.07299, 2018a. URL https://arxiv.org/abs/1801.07299.
- Li et al. (2016) Li, Yujia, Tarlow, Daniel, Brockschmidt, Marc, and Zemel, Richard S. Gated graph sequence neural networks. In Bengio, Yoshua and LeCun, Yann (eds.), 4th International Conference on Learning Representations, ICLR 2016, San Juan, Puerto Rico, May 2-4, 2016, Conference Track Proceedings, 2016. URL http://arxiv.org/abs/1511.05493.
- Li et al. (2018b) Li, Yujia, Vinyals, Oriol, Dyer, Chris, Pascanu, Razvan, and Battaglia, Peter. Learning deep generative models of graphs. ArXiv preprint, abs/1803.03324, 2018b. URL https://arxiv.org/abs/1803.03324.
- Lillicrap et al. (2016) Lillicrap, Timothy P., Hunt, Jonathan J., Pritzel, Alexander, Heess, Nicolas, Erez, Tom, Tassa, Yuval, Silver, David, and Wierstra, Daan. Continuous control with deep reinforcement learning. In Bengio, Yoshua and LeCun, Yann (eds.), 4th International Conference on Learning Representations, ICLR 2016, San Juan, Puerto Rico, May 2-4, 2016, Conference Track Proceedings, 2016. URL http://arxiv.org/abs/1509.02971.
- Maddison et al. (2017) Maddison, Chris J., Mnih, Andriy, and Teh, Yee Whye. The concrete distribution: A continuous relaxation of discrete random variables. In 5th International Conference on Learning Representations, ICLR 2017, Toulon, France, April 24-26, 2017, Conference Track Proceedings. OpenReview.net, 2017. URL https://openreview.net/forum?id=S1jE5L5gl.
- Minervini et al. (2017) Minervini, Pasquale, Demeester, Thomas, Rocktäschel, Tim, and Riedel, Sebastian. Adversarial sets for regularising neural link predictors. In Elidan, Gal, Kersting, Kristian, and Ihler, Alexander T. (eds.), Proceedings of the Thirty-Third Conference on Uncertainty in Artificial Intelligence, UAI 2017, Sydney, Australia, August 11-15, 2017. AUAI Press, 2017. URL http://auai.org/uai2017/proceedings/papers/306.pdf.
- Ramakrishnan et al. (2014) Ramakrishnan, Raghunathan, Dral, Pavlo O, Rupp, Matthias, and Von Lilienfeld, O Anatole. Quantum chemistry structures and properties of 134 kilo molecules. Scientific data, 1:140022, 2014.
- Rezende et al. (2014) Rezende, Danilo Jimenez, Mohamed, Shakir, and Wierstra, Daan. Stochastic backpropagation and approximate inference in deep generative models. In Proceedings of the 31th International Conference on Machine Learning, ICML 2014, Beijing, China, 21-26 June 2014, volume 32 of JMLR Workshop and Conference Proceedings, pp. 1278–1286. JMLR.org, 2014. URL http://proceedings.mlr.press/v32/rezende14.html.
- Ruddigkeit et al. (2012) Ruddigkeit, Lars, Van Deursen, Ruud, Blum, Lorenz C, and Reymond, Jean-Louis. Enumeration of 166 billion organic small molecules in the chemical universe database gdb-17. Journal of chemical information and modeling, 52(11):2864–2875, 2012.
- Salimans et al. (2016) Salimans, Tim, Goodfellow, Ian J., Zaremba, Wojciech, Cheung, Vicki, Radford, Alec, and Chen, Xi. Improved techniques for training gans. In Lee, Daniel D., Sugiyama, Masashi, von Luxburg, Ulrike, Guyon, Isabelle, and Garnett, Roman (eds.), Advances in Neural Information Processing Systems 29: Annual Conference on Neural Information Processing Systems 2016, December 5-10, 2016, Barcelona, Spain, pp. 2226–2234, 2016. URL https://proceedings.neurips.cc/paper/2016/hash/8a3363abe792db2d8761d6403605aeb7-Abstract.html.
- Samanta et al. (2018) Samanta, Bidisha, De, Abir, Ganguly, Niloy, and Gomez-Rodriguez, Manuel. Designing random graph models using variational autoencoders with applications to chemical design. ArXiv preprint, abs/1802.05283, 2018. URL https://arxiv.org/abs/1802.05283.
- Schlichtkrull et al. (2017) Schlichtkrull, Michael, Kipf, Thomas N, Bloem, Peter, Berg, Rianne van den, Titov, Ivan, and Welling, Max. Modeling relational data with graph convolutional networks. ArXiv preprint, abs/1703.06103, 2017. URL https://arxiv.org/abs/1703.06103.
- Schneider & Fechner (2005) Schneider, Gisbert and Fechner, Uli. Computer-based de novo design of drug-like molecules. Nature Reviews Drug Discovery, 4(8):649, 2005.
- Segler et al. (2018) Segler, Marwin HS, Preuss, Mike, and Waller, Mark P. Planning chemical syntheses with deep neural networks and symbolic ai. Nature, 555(7698):604, 2018.
- Silver et al. (2014) Silver, David, Lever, Guy, Heess, Nicolas, Degris, Thomas, Wierstra, Daan, and Riedmiller, Martin A. Deterministic policy gradient algorithms. In Proceedings of the 31th International Conference on Machine Learning, ICML 2014, Beijing, China, 21-26 June 2014, volume 32 of JMLR Workshop and Conference Proceedings, pp. 387–395. JMLR.org, 2014. URL http://proceedings.mlr.press/v32/silver14.html.
- Simonovsky & Komodakis (2018) Simonovsky, Martin and Komodakis, Nikos. Graphvae: Towards generation of small graphs using variational autoencoders. ArXiv preprint, abs/1802.03480, 2018. URL https://arxiv.org/abs/1802.03480.
- Srivastava et al. (2014) Srivastava, Nitish, Hinton, Geoffrey, Krizhevsky, Alex, Sutskever, Ilya, and Salakhutdinov, Ruslan. Dropout: A simple way to prevent neural networks from overfitting. The Journal of Machine Learning Research, 15(1):1929–1958, 2014.
- Wang et al. (2018) Wang, Hongwei, Wang, Jia, Wang, Jialin, Zhao, Miao, Zhang, Weinan, Zhang, Fuzheng, Xie, Xing, and Guo, Minyi. Graphgan: Graph representation learning with generative adversarial nets. In McIlraith, Sheila A. and Weinberger, Kilian Q. (eds.), Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, (AAAI-18), the 30th innovative Applications of Artificial Intelligence (IAAI-18), and the 8th AAAI Symposium on Educational Advances in Artificial Intelligence (EAAI-18), New Orleans, Louisiana, USA, February 2-7, 2018, pp. 2508–2515. AAAI Press, 2018. URL https://www.aaai.org/ocs/index.php/AAAI/AAAI18/paper/view/16611.
- Weininger (1988) Weininger, David. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. Journal of chemical information and computer sciences, 28(1):31–36, 1988.
- Williams (1992) Williams, Ronald J. Simple statistical gradient-following algorithms for connectionist reinforcement learning. In Reinforcement Learning, pp. 5–32. Springer, 1992.
- You et al. (2018) You, Jiaxuan, Ying, Rex, Ren, Xiang, Hamilton, William L, and Leskovec, Jure. Graphrnn: A deep generative model for graphs. In International Conference on Machine Learning, 2018.
- Yu et al. (2017) Yu, Lantao, Zhang, Weinan, Wang, Jun, and Yu, Yong. Seqgan: Sequence generative adversarial nets with policy gradient. In Singh, Satinder P. and Markovitch, Shaul (eds.), Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence, February 4-9, 2017, San Francisco, California, USA, pp. 2852–2858. AAAI Press, 2017. URL http://aaai.org/ocs/index.php/AAAI/AAAI17/paper/view/14344.