Grappa - 机器学习的分子力学力场
摘要
长时间模拟大型分子系统需要准确且高效的力场。 近年来,E(3) 等变神经网络缓解了计算效率和力场精度之间的紧张关系,但它们仍然比经典分子力学 (MM) 力场昂贵几个数量级。 在这里,我们提出了一种新颖的机器学习架构,利用图注意力神经网络和具有保对称位置编码的 Transformer 从分子图预测 MM 参数。 由此产生的力场 Grappa 在相同计算效率的精度方面优于现有的和其他机器学习的 MM 力场,并且可用于现有的分子动力学 (MD) 引擎,例如 GROMACS 和 OpenMM。 它以最先进的 MM 精度预测小分子、肽、RNA 和自由基的能量和作用力(展示其对化学空间未知区域的扩展性)。 我们在 MD 模拟中证明了 Grappa 对大分子的可转移性,在此期间大蛋白质保持稳定,小蛋白质可以折叠。 我们的力场为接近化学精度的生物分子模拟奠定了基础,但计算成本与已建立的蛋白质力场相同。
1简介
机器学习力场目前正在重塑计算化学和分子动力学 (MD) 模拟领域。 E(3)-等变神经网络[1,2,3,4,5]能够以比量子力学(QM)方法更低的计算成本高精度预测小分子的能量和力。 然而,这些模型比分子力学 (MM) 力场昂贵几个数量级,分子力学 (MM) 力场采用简单的受物理启发的函数形式来参数化分子系统的势能表面,因此需要牺牲准确性以提高效率。 对于蛋白质和核酸等大型系统的 MD 模拟,MM 力场已经成熟并广泛使用。 传统的 MM 力场依赖于具有一组有限原子类别的查找表,这些原子类别描述了局部化学环境以进行参数化。 最近,Espaloma 方法[6] 证明,机器学习可以通过学习基于具有手工制作的化学特征的分子的图形表示来分配参数,从而提高 MM 力场的准确性例如轨道杂化态。
在这项工作中,我们提出了一种新颖的机器学习架构 Grappa(图注意蛋白参数化),直接从分子图学习 MM 参数,提高了广泛化学空间的准确性,并消除了手工制作特征的需要。 Grappa 采用图注意力神经网络来预测局部化学环境,然后使用具有对称性保持位置编码的 Transformer [7]。 由于每个分子只需预测一次 MM 参数,因此 Grappa 可以合并到高度优化的 MM 引擎中,例如 GROMACS [8] 和 OpenMM [9]。 这允许以与传统力场相同的计算成本和最先进的 MM 精度来评估能量和力。
我们在 Espaloma 数据集[10]上证明 Grappa 优于传统 MM 力场和 Espaloma,该数据集包含超过 14,000 个分子和超过 100 万个状态,涵盖小分子、肽和 RNA。 由于 Grappa 不使用手工制作的化学特征,因此它可以扩展到化学空间的未知区域,我们在肽自由基的例子中证明了这一点。 Grappa 可转移至蛋白质等大分子,并在纳秒时间尺度的 MD 过程中保持稳定。 从展开的初始状态开始,Grappa 参数化的小蛋白质的 MD 模拟恢复了实验确定的小蛋白质的折叠结构,这表明 Grappa 捕获了蛋白质折叠背后的物理原理。
1.1分子力学
给定分子图,MM 将系统的势能表示为不同相互作用的贡献之和。 键合相互作用通过 E(3) 不变内坐标的函数来描述,例如两个原子之间键的长度 、三个连续原子之间的角度 和二面体 由四个原子跨越的两个平面。 对于二面角,人们考虑四个原子之间的相互作用,这些原子要么连续键合(扭转),要么三个原子键合到一个中心原子(不正确),这并不反映独立的自由度,而是用于维持某些化学物质的平面性组。 人们通常使用调和势来进行键拉伸和角度弯曲,并使用周期函数来计算二面角势。 沿键的所有相互作用的势能由下式给出
| (1) |
平衡值(键和角) 和 ,以及力常数 、 和 ,作为 MM 参数集,我们表示为
| (2) |
对于 Grappa 中的二面势,我们选择傅立叶级数,其约束条件是二面势在零处为极值且在零附近对称,这样就不需要正弦项。 此外,不包含在此类 N 体键合相互作用项中的原子对通过成对非键合相互作用贡献势能,通常由 Lennard-Jones 和库仑势描述。
传统的MM力场定义了由手工制定的规则确定的有限原子类型集,这些规则用于根据原子类型可能的组合的查找表来分配自由参数。 在 Grappa 中,我们通过直接从分子图中学习参数来取代这种方案,这使得势能面的描述更加灵活和可转移。
标准 MM 的一个基本限制是恒定分子图拓扑的假设,这是通过使用谐波键势来强制执行的。 虽然这限制了准确性并禁止描述键变化化学反应,但势能函数的物理可解释性确保模拟系统保持稳定,即使在力场难以描述的状态下也是如此。
2格拉巴酒
受到传统 MM 力场中手工制定规则的原子打字的启发,并类似于 [6],Grappa 首先预测 维原子嵌入,
| (3) |
它可以代表分子图结构中编码的局部化学环境,其中节点集代表原子,边集 代表债券。 在第二步(图1)中,对于每种相互作用类型,MM参数是根据参与各自能量贡献的原子的嵌入来预测的,
| (4) |
使用在某些排列下不变的 Transformer 。 利用MM的能量函数,预测参数集定义了一个势能面,最终可以针对分子的不同空间构象进行评估,
| (5) |
由于从分子图到能量的映射对于模型参数和空间位置是可微的,因此可以在端到端预测 QM 能量和力方面进行优化。 值得注意的是,机器学习模型不依赖于分子的空间构象,因此每个分子只需评估一次,并且每次后续能量评估的计算成本由 MM 能量函数给出。
Grappa 目前仅预测键合 MM 参数,因为非键合相互作用的高度非线性能量函数阻碍了优化。 非键合参数取自已建立的 MM 力场,该力场可以重现溶质相互作用和熔点,我们预计它们强烈依赖于非键合相互作用。
2.1 MM中的排列对称性
对于从原子嵌入 到 MM 参数的映射,我们假设我们的模型应该遵循某些排列对称性。 为了推导这些对称性,我们将 MM 的能量函数视为分解为特征化分子图的子图的贡献,这些子图对应于键、角度、扭转和不正确的二面角。 我们要求节点排列下能量贡献的不变性,从而引起相应子图的同构。 对于键、角度和扭转,这些排列使各自的空间坐标保持不变,因此我们可以通过要求 MM 参数的不变性来简单地实现能量贡献的不变性,
| (6) | ||||
| (7) | ||||
| (8) |
然而,对于不正确的二面角,并非所有子图同构都使二面角保持不变,并且在这些排列下要求参数不变性将导致能量贡献不不变。 在 Grappa 中,我们通过将不正确的扭转贡献分解为三项来解决这个问题,如 A.4 中所述。
2.2 Grappa架构
为了从分子图中预测原子嵌入,Grappa 采用了受 Transformer 架构 [7] 启发的图注意力神经网络。 图边 [11] 上的多头点积注意力后面是节点前馈层。 我们使用残差连接[12]和层归一化[13],这已被证明可以增强注意力层[14]的表达能力。
对于从这些嵌入到 MM 参数的映射,最好使用尊重排列对称性的架构(等式 1)。 6 - 8)通过设计,将可能模型的空间限制为物理上合理的模型。 本着等变机器学习的精神,我们使用排列等变层,然后是最终的对称池。 然而,由于我们不需要所有排列下的不变性,而只需要在方程式中定义的排列下不变。 6 - 8,我们可以通过允许模型打破不需要的对称性来提高表现力。
考虑到这些因素,我们使用基于多头点注意力的 Transformer 架构,其位置编码在所需的对称性下保持不变,但可能会破坏其他对称性。 例如对于角度,该位置编码由下式给出
| (9) |
其中 操作将相应的值附加到节点特征向量,使其在 下不变,但在例如。 在这些等变层之后,我们在连接的排列节点嵌入上应用多层感知器(MLP),并对所需的排列集 求和,
| (10) |
定义具有 不变输出的对称池操作。 我们将这种排列不变位置编码与排列等变层和对称池的组合称为对称变换器,如图2所示。 通过使用图拉普拉斯[15]的特征向量作为位置编码,对称Transformer可以推广到任意子图的排列对称性。
最后,我们映射到物理上敏感参数的范围,例如 用于键力和角力常数,或 用于平衡角 。 为此,我们使用 ELU 的缩放和移位版本以及 sigmoid 函数,如 A.2.3 中所述。 虽然也可以使用指数来映射到 ,但 ELU 对大输入的线性行为有利于在优化过程中产生稳定的梯度。 通过缩放,我们确保神经网络的正态分布输出映射到适合各自 MM 参数的均值和标准差的分布,这可以看作是一种归一化技术[13]。 为了预测二面体参数 ,我们使用 sigmoid 门,它允许模型抑制不需要的二面体模式。
作为输入特征,我们使用原子序数、各个节点的邻居数量以及长度为 3 到 8 的循环中的成员资格的 one-hot 编码,这些可以直接从分子图计算出来。 非键合 MM 参数的分配是使用选择的传统力场完成的。 由于 Grappa 预测的最佳键合参数可能取决于分配非键合参数的方案,因此我们将每个原子的部分电荷的正弦编码 [7] 作为 Grappa 的输入特征,这也允许对分子的总电荷进行编码,而不会破坏图形对称性,就像使用形式电荷可能会做的那样。 对于 3.3 节中蛋白质的扩展,我们训练 Grappa 与两种不同的非键参数分配方案兼容,我们将其编码为附加输入特征。 这些方案是 a) 我们所说的 AM1-BCC 方案,其中 Lennard-Jones 参数来自 OpenFF-2.0.0 力场,部分电荷来自半经验方法 AM1-BCC [16] b) 我们所说的 ff99SB 方案,具有 Lennard-Jones 参数和来自已建立的 Amber ff99SB-ILDN [17] 蛋白质力场的部分电荷。
2.3训练
我们训练 Grappa 来最小化 QM 能量 和力 之间的均方误差 (MSE),我们通过超参数 及其预测来衡量其贡献、 和 分别,其中包括不可学习的非绑定贡献。 在训练开始时,我们还包括预测的粘合、角度和扭转 MM 参数与给定传统力场的偏差,按 加权,并且为了正则化,使用 二面体 MM 参数范数 ,按 加权,如 [10] 中。 也就是说,我们使用损失函数
| (11) |
由于 MM 只能预测态的能量差,而不能预测形成能,因此我们减去每个分子的目标能量和预测能量的平均值。 如之前针对其他机器学习力场 [18, 5] 所示,我们发现除了能量之外,力 的训练在我们的设置中也很重要,原因有两个。 首先,能量是一个全局的、集中的数量,因此不如力具有表达力,而力是局部的,而且数量多出 倍。 其次,如[19]所示,学习目标相对于输入的导数可以提高泛化能力和数据效率,有效地平滑学习到的势能面。
3 结果
3.1 基准测试结果
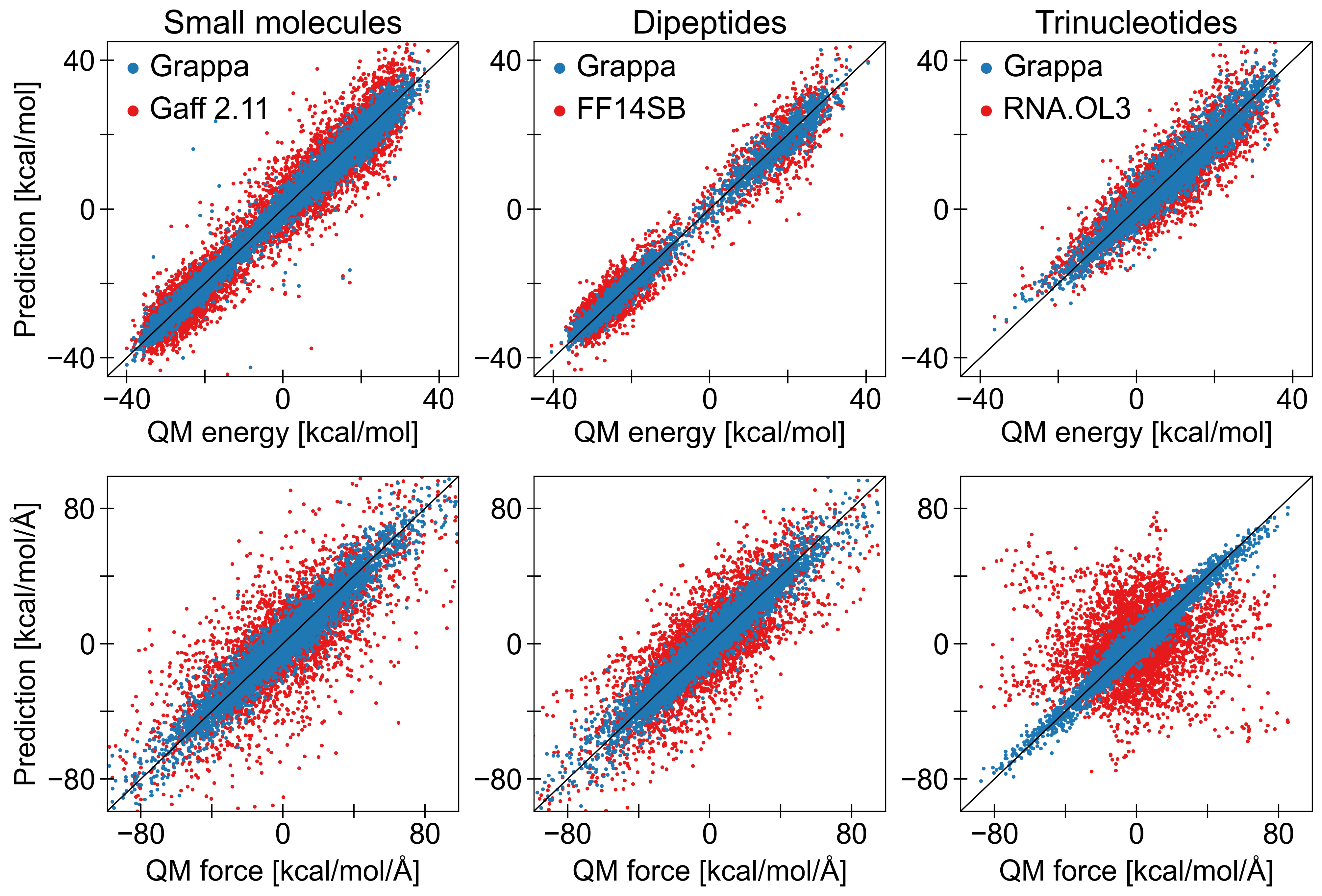
为了证明 Grappa 是当前最先进的 MM 力场,我们在 Espaloma 的后续论文 [10] 中报告的数据集上训练并评估 Grappa 模型,该数据集包含 17,427 个独特的分子和超过一百万个构象。 我们的训练和测试分区与 Espaloma 的训练和测试分区相同,其中分子根据异构 SMILES 字符串分为 80% 训练集、10% 验证集和 10% 测试集。 该数据集涵盖小分子、肽和 RNA,其状态是从 300 K 和 500 K 的玻尔兹曼分布、优化轨迹和扭转扫描中采样的。 非键合贡献是使用 OpenFF-2.0.0 力场 [20] 和 AM1-BCC 方法的部分电荷计算的,如 Espaloma 中所示。
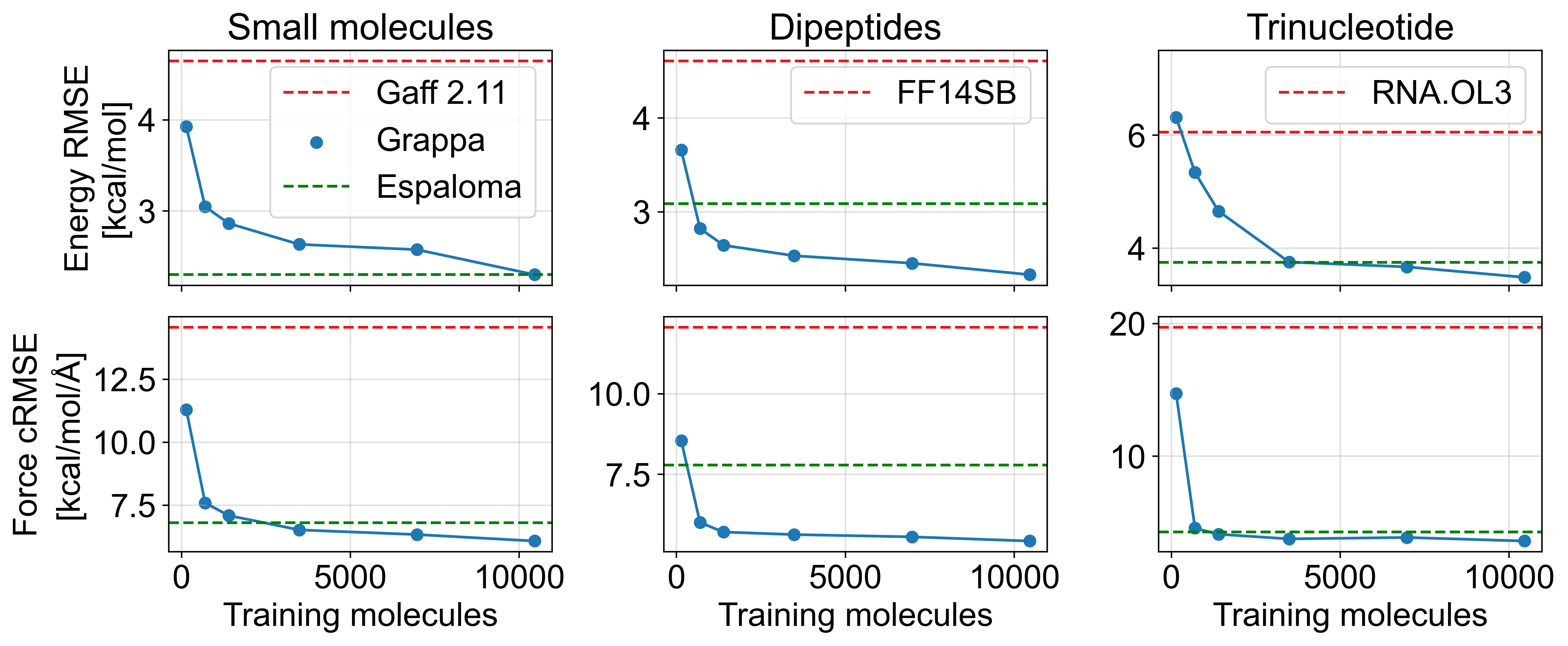
对于所有类型的分子,Grappa 在玻尔兹曼采样态的能量和力精度方面优于已建立的 MM 力场和 Espaloma,如表 1 和图 3a 所示。 虽然 Boltzmann 样本是在 MD 模拟中使用 Grappa 的更相关的基准数据,但我们还比较了扭转扫描和优化轨迹的性能(表 A4)。 在那里,我们发现 Grappa 和 Espaloma 具有竞争力,而 Grappa 对于力而言更准确,而对于能量而言则不太准确。 如果 Grappa 仅在 Espaloma 训练集的一小部分上进行训练(图A9),情况也是如此,表明数据效率很高。
| Dataset | Test Mols | Confs | Grappa | Espaloma | Gaff-2.11 | ff14SB, | Mean | |
|---|---|---|---|---|---|---|---|---|
| RNA.OL3 | Predictor | |||||||
| SPICE-Pubchem | 1411 | 60853 | Energy | 2.3 | 2.3 | 4.6 | 18.4 | |
| Force | 6.1 | 6.8 | 14.6 | 23.4 | ||||
| SPICE-DES-Monomers | 39 | 2032 | Energy | 1.3 | 1.4 | 2.5 | 8.2 | |
| Force | 5.2 | 5.9 | 11.1 | 21.3 | ||||
| SPICE-Dipeptide | 67 | 2592 | Energy | 2.3 | 3.1 | 4.5 | 4.6 | 18.7 |
| Force | 5.4 | 7.8 | 12.9 | 12.1 | 21.6 | |||
| RNA-Diverse | 6 | 357 | Energy | 3.3 | 4.2 | 6.5 | 6.0 | 5.4 |
| Force | 3.7 | 4.4 | 16.7 | 19.4 | 17.1 | |||
| RNA-Trinucleotide | 64 | 35811 | Energy | 3.5 | 3.8 | 5.9 | 6.1 | 5.3 |
| Force | 3.6 | 4.3 | 17.1 | 19.7 | 17.7 |
3.2 潜在空间
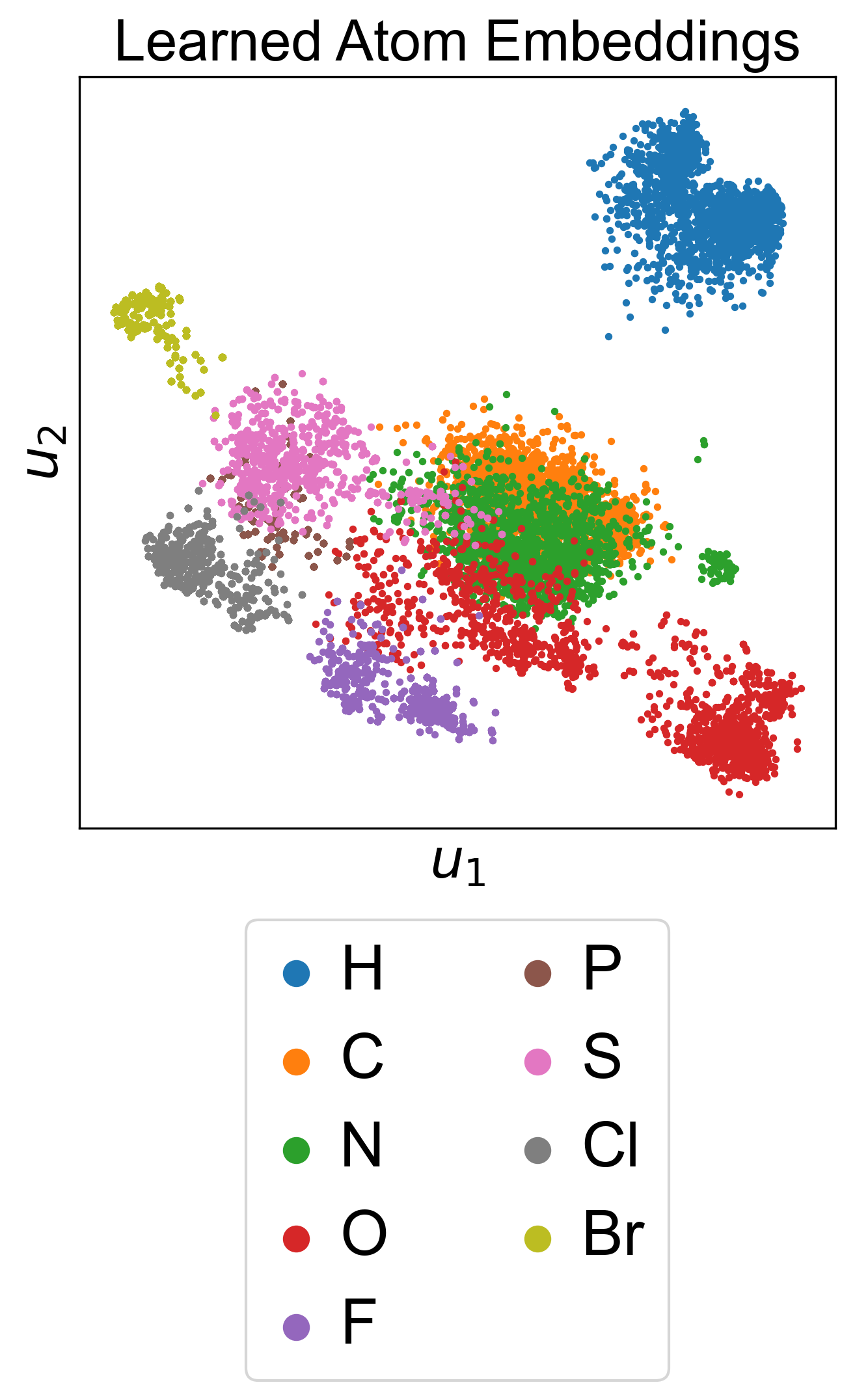
3.3 扩展到蛋白质
| Dataset | Test Mols | Confs | Grappa | Grappa | Espaloma | ff14SB, | Mean | |
|---|---|---|---|---|---|---|---|---|
| AM1-BCC | ff99SB | RNA.OL3 | Predictor | |||||
| SPICE-Dipeptide | 67 | 2592 | Energy | 2.3 | 2.2 | 3.1 | 4.6 | 4.3 |
| Force | 5.5 | 5.9 | 7.8 | 12.1 | 10.7 | |||
| Radical-Dipeptide | 28 | 275 | Energy | 2.8 | 7.4 | |||
| Force | 6.5 | 23.6 | ||||||
| HYP-DOP-Dipeptide | 8 | 363 | Energy | 2.3 | 6.8 | |||
| Force | 5.6 | 24.9 | ||||||
| Uncapped-Dipeptide | 12 | 600 | Energy | 2.1 | 3.7 | 6.5 | ||
| Force | 5.9 | 11.7 | 25.3 |
特别是对于大的生物分子,力场不仅需要尽可能准确地预测能量和力,而且它们的预测应该以这样的方式进行:在MD模拟中满足某些宏观特性,例如稳定性和与实验确定的折叠状态的一致性。 Grappa 非常适合克服经经验验证的、已建立的蛋白质力场和机器学习力场之间的差距。
我们表明,可以训练 Grappa 来预测与来自已建立的 Amber ff99SB-ILDN 力场 [17] 和 AM1-BCC 方法的非键合参数一致的键合参数。 这使得能够在使用已建立的非键合参数的同时准确预测能量和力,我们预计这些参数将主要负责稳定性和折叠状态等特性。 由于 Grappa 不依赖于手工制作的化学输入功能,因此将其纳入现有的生物分子 MD 引擎(如 GROMACS [8])非常简单,并且其工作流程可以扩展到大分子;仅需要连通性和原子序数作为附加输入特征。 通过超参数 及其 MM 公式,它还提供了在已建立的力场和完全机器学习的力场之间平滑且一致地进行插值的选项。
为了使 Grappa 适用于蛋白质,我们在 Grappa-1.1 数据集上进行训练,该数据集是 Espaloma 数据集的扩展,包含非标准残基羟脯氨酸和 DOPA(二羟基苯丙氨酸)、无帽肽,并证明了 Grappa 的参数化能力不常见的分子、肽基团,均具有来自 Amber ff99SB-ILDN 的非键合参数。 这一选择的动机是 Grappa 未来在胶原蛋白和自由基中的应用,但这里是为了证明 Grappa 可以轻松扩展到标准生物分子力场之外的化学领域。 为了创建这些额外的数据集,我们计算了从使用 300K 的传统力场进行的 MD 模拟中采样的状态的 DFT 能量和梯度,其中我们在采样之间将温度提高到 500 K 以丰富构象多样性,如 A.5。
4 强制字段验证
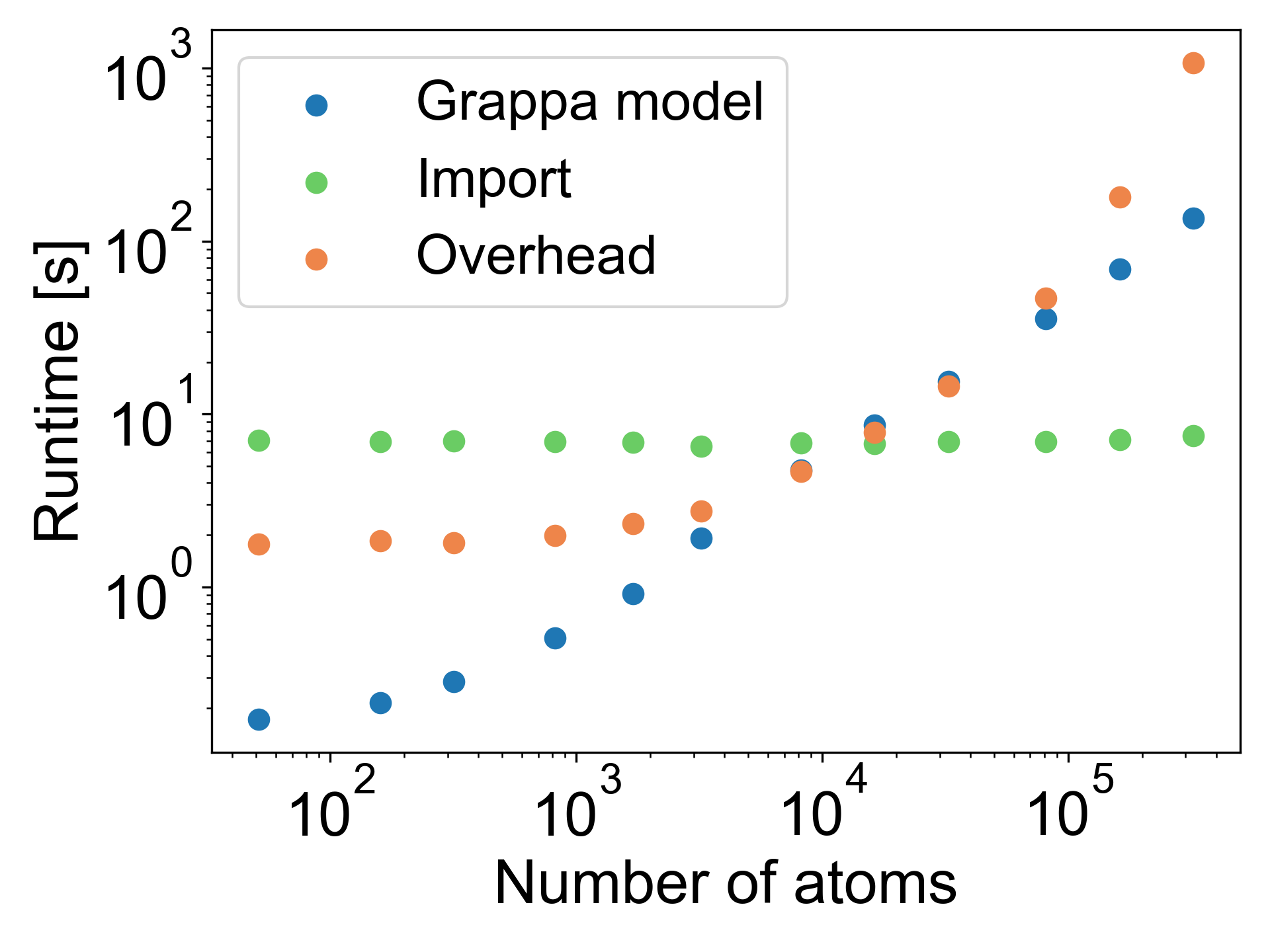
在本节中,我们将证明 Grappa 在数百纳秒的时间尺度上对蛋白质进行 MD 模拟时与已建立的力场相当,并且它捕获了蛋白质折叠的物理原理。 本节提到的所有模拟均使用 GROMACS 执行,Grappa 软件包111https://github.com/hits-mbm-dev/grappa提供了一个命令行界面,在A.1中进行了简要描述。 使用此接口(或 OpenMM 的类似接口),具有超过一万个原子的大型生物分子系统可在几分钟内在 CPU 上参数化(图 A6)。
4.1 Grappa 在 MD 过程中保持大蛋白稳定
蛋白质力场的关键质量检查是天然蛋白质结构在 MD 模拟过程中是否随时间保持稳定。 QM 方法和 E(3) 等变神经网络存在这样的问题:虽然它们的端到端预测是准确的,但由于在外推到训练覆盖的区域之外时,势能表面的梯度发散,它们常常无法产生稳定的模拟。数据集。 特别是在较长的时间尺度上,系统可能会偏离模型的有效性范围,并且小错误可能会累积,从而导致系统不稳定并可能导致模拟崩溃。 MM 具有可解释的、受物理启发的能量函数以及与构象无关的参数,确保了更高程度的稳定性,而且我们确实没有观察到模拟崩溃。
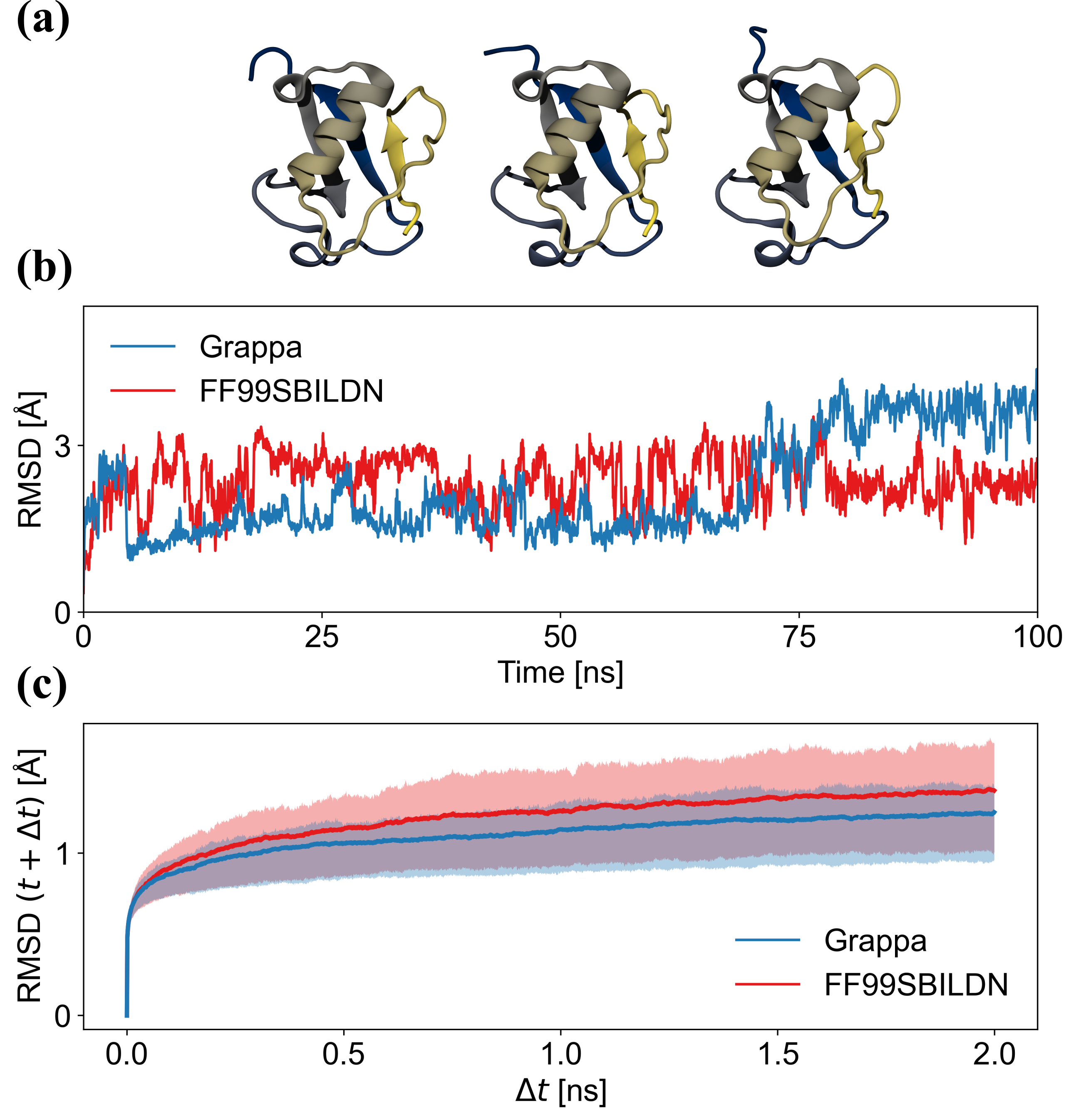
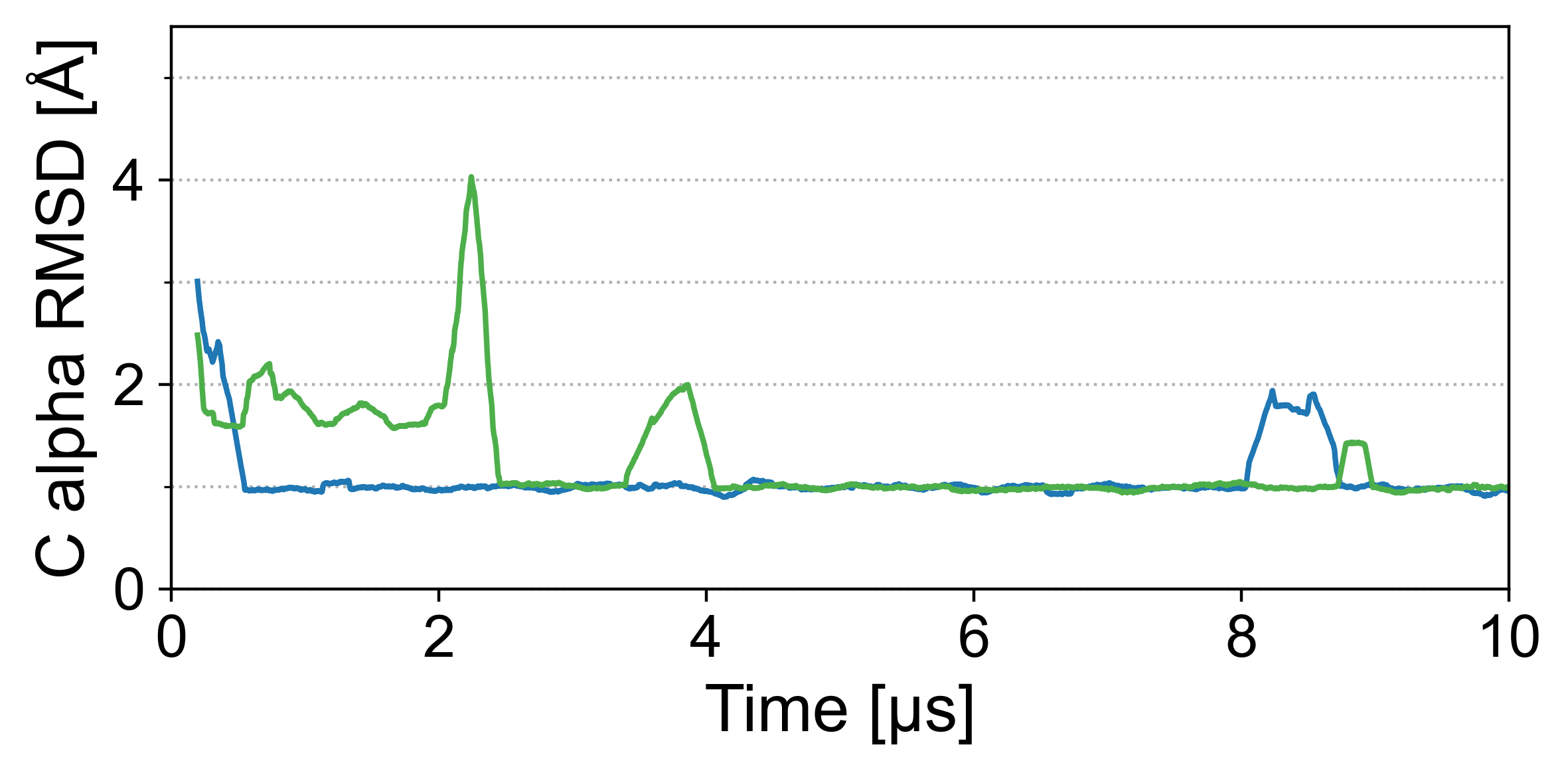
然而,显然,不能先验地确保蛋白质折叠等宏观状态在 MD 过程中保持稳定。 接下来我们评估 Grappa 保持蛋白质折叠接近其实验确定的结构的能力。 作为示例系统,我们使用泛素(PDB ID:1UBQ),它是一种具有 76 个氨基酸的蛋白质,其折叠包含 β 折叠和 α 螺旋。 我们使用 Grappa 和 Amber ff99SB-ILDN [17] 对水溶液中的泛素进行 100 纳秒 MD 模拟,详细信息参见 A.6.2。 我们发现,在 Grappa 和 Amber ff99SB-ILDN 的 100 ns 模拟中,初始状态的 C-alpha RMSD 以 4 Å 为界(图4b),表明蛋白质的折叠状态是稳定的。 Grappa 的时间尺度上的结构波动更小,可达 2 ns(图4c)。
4.2 Grappa可以通过MD折叠小蛋白质
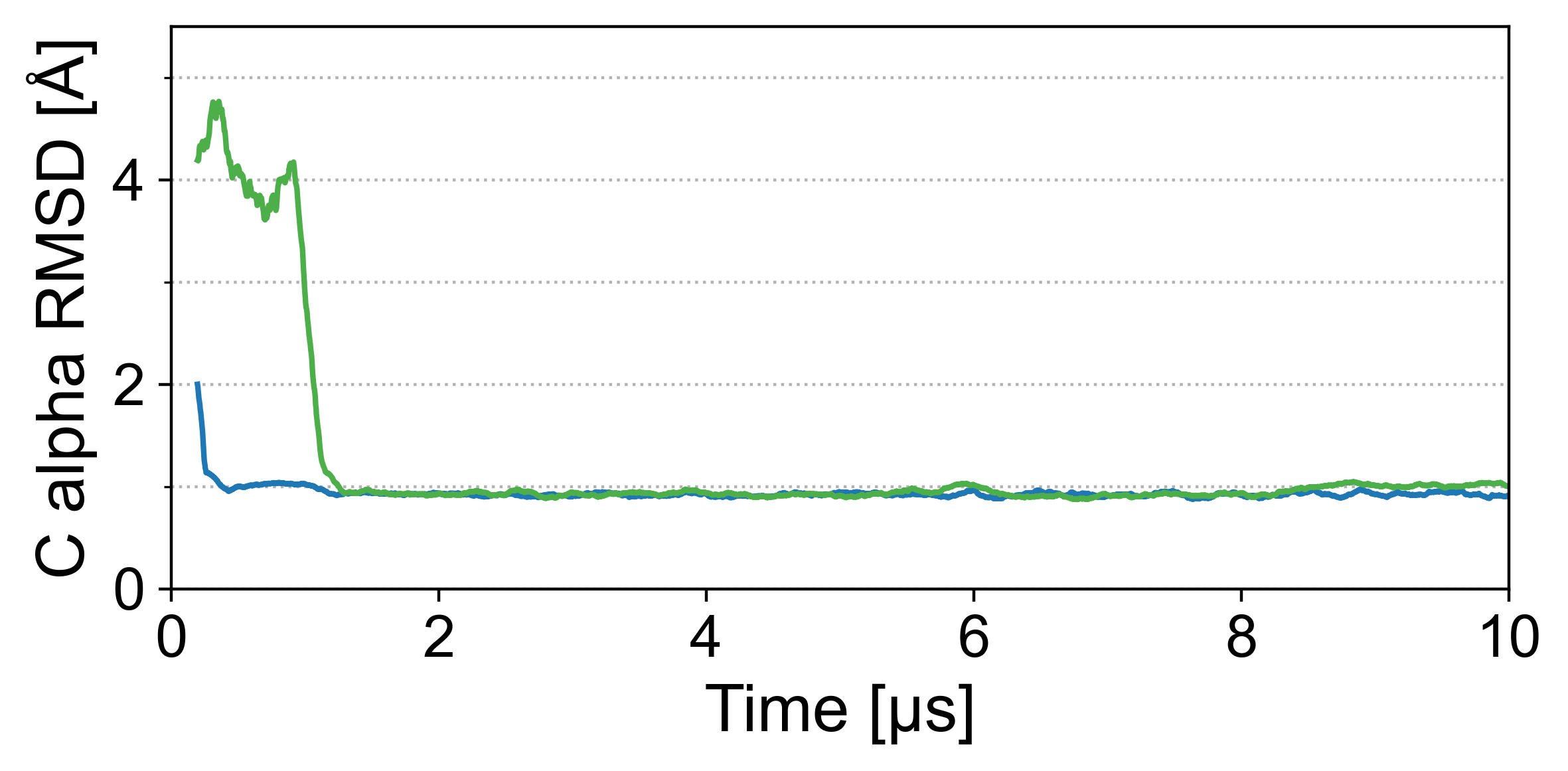
为了评估 Grappa 对构象空间的覆盖范围,甚至远离折叠蛋白质状态,我们研究了 chignolin(一种含有 10 个氨基酸的小蛋白质)的折叠轨迹。 从展开状态开始,我们使用 Grappa 和 Amber ff99SB-ILDN [17] 对水溶液中的 chignolin 进行两次 10 微秒 MD 模拟,如 A.6.1 中所述。 对于后者,已经证明它可以在类似的设置中折叠 chignolin (Lindorff-Larsen 等人 [25]),其中人口最多的簇的 C-alpha RMSD 为 1 Å到实验确定的参考结构(PDB ID:5AWL)。
我们发现 Grappa 在模拟微秒内将 chignolin 折叠成实验确定的结构,如图 5 所示。 填充最多的簇与参考结构的 RMSD 为 0.8 Å,而 Amber ff99SB-ILDN 模拟中填充最多的簇的 RMSD 为 0.9 Å,两者均小于参考结构的分辨率 (1.1 Å)。 Grappa 的折叠轨迹在质量上与 Amber ff99SB-ILDN 的折叠轨迹相似,证实了 Grappa 对于蛋白质模拟的有效性。


5结论
通过 Grappa,我们提出了一个机器学习框架,以最先进的精度构建分子力学力场,可以在已建立的 MD 引擎中无缝使用。 我们的实验表明,Grappa 可转移到蛋白质等大型生物分子,这为蛋白质模拟提供了前所未有的精确度,其计算效率与已建立的 MM 力场相同,但误差减少了两倍。
与大多数传统的 MM 力场不同,Grappa 并不专门针对某种化学物质,而是通过单一模型对化学空间不同区域的分子提供一致的参数化。 我们预计 Grappa 是一个通用且多功能的框架,可以根据需要将可用参数集扩展到新的化学物质,例如药物、不寻常的脂质、复杂的糖或稀有的蛋白质修饰。
虽然 Grappa 通过分子力学实现了高计算效率,但它也继承了其基本局限性,包括有限的精度和无法直接模拟化学反应。 然而,Grappa 非常适合对经历由化学反应引起的局部变化的大分子进行有效的重新参数化,例如在动力学蒙特卡罗模拟中[26]。 这是由于 Grappa 的有限视场(确保了分子图上的局部性)以及缺乏手工制作的化学特征(允许将参数化应用于分子的剪切区域)。
虽然我们认为与已建立的非键参数的一致性是 Grappa 的优势,但下一步是将我们的框架扩展到非键参数。 它将使 Grappa 在推理时完全独立于传统的 MM 力场,并有可能进一步提高其准确性。
6 再现性声明
Grappa 作为开源软件根据 GNU 通用公共许可证 v3.0 发布,可从 https://github.com/hits-mbm-dev/grappa 获取。 该存储库包含 Grappa 数据集以及用于 train-val-test 分区的 SMILES 字符串以及用于重现表 1 和表 2 的配置文件。
折叠和稳定性模拟可以使用 https://github.com/LeifSeute/validate-grappa 上的存储库进行重现,使用无帽肽的数据集的创建记录在 https:// github.com/LeifSeute/grappa-data-creation。
Grappa 在其实现中使用 PyTorch [27] 和 DGL [28] 。
作者贡献
莱夫·苏特: 方法论、概念化、软件、写作 - 初稿、调查、可视化、数据管理
埃里克·哈特曼: 概念化、软件、验证、可视化、写作 - 审查和编辑
扬·斯图默: 形式分析、写作-评论和编辑
弗劳克·格瑞特: 监督、概念化、写作 - 审查和编辑
利益冲突
没有需要声明的冲突。
致谢
这项研究得到了克劳斯·奇拉基金会的支持。 该项目还获得了欧洲研究理事会(ERC)根据欧盟地平线 2020 研究和创新计划提供的资助(资助协议号: 101002812)[F.G.]。 我们感谢 Kai Riedmiller 和 Jannik Buhr 进行应用程序和软件相关的讨论,感谢 Fabian Grünewald 和 Camilo Aponte-Santamaria 进行关于未来应用的讨论,感谢 Daniel Sucerquia 进行关于 MM 中内坐标作用的对话。
参考
- Musaelian et al. [2022] Albert Musaelian, Simon Batzner, Anders Johansson, Lixin Sun, Cameron J. Owen, Mordechai Kornbluth, and Boris Kozinsky. Learning local equivariant representations for large-scale atomistic dynamics, 2022. URL https://arxiv.org/abs/2204.05249.
- Batatia et al. [2023] Ilyes Batatia, Dávid Péter Kovács, Gregor N. C. Simm, Christoph Ortner, and Gábor Csányi. Mace: Higher order equivariant message passing neural networks for fast and accurate force fields, 2023.
- Simeon and de Fabritiis [2023] Guillem Simeon and Gianni de Fabritiis. Tensornet: Cartesian tensor representations for efficient learning of molecular potentials, 2023.
- Unke et al. [2022] Oliver T. Unke, Martin Stöhr, Stefan Ganscha, Thomas Unterthiner, Hartmut Maennel, Sergii Kashubin, Daniel Ahlin, Michael Gastegger, Leonardo Medrano Sandonas, Alexandre Tkatchenko, and Klaus-Robert Müller. Accurate machine learned quantum-mechanical force fields for biomolecular simulations, 2022.
- Schütt et al. [2021] Kristof T. Schütt, Oliver T. Unke, and Michael Gastegger. Equivariant message passing for the prediction of tensorial properties and molecular spectra, 2021. URL https://arxiv.org/abs/2102.03150.
- Wang et al. [2022] Yuanqing Wang, Josh Fass, Benjamin Kaminow, John E. Herr, Dominic Rufa, Ivy Zhang, Ivá n Pulido, Mike Henry, Hannah E. Bruce Macdonald, Kenichiro Takaba, and John D. Chodera. End-to-end differentiable construction of molecular mechanics force fields. Chemical Science, 13(41):12016–12033, 2022. doi: 10.1039/d2sc02739a. URL https://doi.org/10.1039%2Fd2sc02739a.
- Vaswani et al. [2023] Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N. Gomez, Lukasz Kaiser, and Illia Polosukhin. Attention is all you need, 2023. URL https://arxiv.org/abs/1706.03762.
- Abraham et al. [2015] Mark James Abraham, Teemu Murtola, Roland Schulz, Szilárd Páll, Jeremy C. Smith, Berk Hess, and Erik Lindahl. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1-2:19–25, 2015. ISSN 2352-7110.
- Eastman et al. [2013] Peter Eastman, Mark S. Friedrichs, John D. Chodera, Randall J. Radmer, Christopher M. Bruns, Joy P. Ku, Kyle A. Beauchamp, Thomas J. Lane, Lee-Ping Wang, Diwakar Shukla, Tony Tye, Mike Houston, Timo Stich, Christoph Klein, Michael R. Shirts, and Vijay S. Pande. Openmm 4: A reusable, extensible, hardware independent library for high performance molecular simulation. Journal of Chemical Theory and Computation, 9(1):461–469, 2013. doi: 10.1021/ct300857j. URL https://doi.org/10.1021/ct300857j. PMID: 23316124.
- Takaba et al. [2023] Kenichiro Takaba, Iván Pulido, Mike Henry, Hugo MacDermott-Opeskin, John D. Chodera, and Yuanqing Wang. Espaloma-0.3.0: Machine-learned molecular mechanics force field for the simulation of protein-ligand systems and beyond, 2023.
- Veličković et al. [2018] Petar Veličković, Guillem Cucurull, Arantxa Casanova, Adriana Romero, Pietro Liò, and Yoshua Bengio. Graph attention networks, 2018. URL https://arxiv.org/abs/1710.10903.
- He et al. [2015] Kaiming He, Xiangyu Zhang, Shaoqing Ren, and Jian Sun. Deep residual learning for image recognition, 2015.
- Ba et al. [2016] Jimmy Lei Ba, Jamie Ryan Kiros, and Geoffrey E. Hinton. Layer normalization, 2016.
- Brody et al. [2023] Shaked Brody, Uri Alon, and Eran Yahav. On the expressivity role of layernorm in transformers’ attention. arXiv preprint arXiv:2305.02582, 2023.
- Yun et al. [2020] Seongjun Yun, Minbyul Jeong, Raehyun Kim, Jaewoo Kang, and Hyunwoo J. Kim. Graph transformer networks, 2020.
- Jakalian et al. [2002] Araz Jakalian, David B. Jack, and Christopher I. Bayly. Fast, efficient generation of high-quality atomic charges. am1-bcc model: Ii. parameterization and validation. Journal of Computational Chemistry, 23(16):1623–1641, 2002. doi: https://doi.org/10.1002/jcc.10128. URL https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.10128.
- Lindorff-Larsen et al. [2010] K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J.L. Klepeis, R.O. Dror, and D.E. Shaw. Improved side-chain torsion potentials for the amber ff99sb protein force field. Proteins, 78(8):1950–1958, Jun 2010. doi: 10.1002/prot.22711. URL https://doi.org/10.1002/prot.22711.
- Schütt et al. [2017] Kristof T. Schütt, Pieter-Jan Kindermans, Huziel E. Sauceda, Stefan Chmiela, Alexandre Tkatchenko, and Klaus-Robert Müller. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions, 2017. URL https://arxiv.org/abs/1706.08566.
- Czarnecki et al. [2017] Wojciech Marian Czarnecki, Simon Osindero, Max Jaderberg, Grzegorz Świrszcz, and Razvan Pascanu. Sobolev training for neural networks, 2017.
- Boothroyd et al. [2022] S. Boothroyd, P.K. Behara, O. Madin, D. Hahn, H. Jang, V. Gapsys, et al. Development and benchmarking of open force field 2.0.0 — the sage small molecule force field. ChemRxiv, 2022. doi: 10.1021/acs.jctc.3c00039. URL https://doi.org/10.1021/acs.jctc.3c00039. This content is a preprint and has not been peer-reviewed.
- He et al. [2020] Xibing He, Viet Man, Wei Yang, Tai-Sung Lee, and Junmei Wang. A fast and high-quality charge model for the next generation general amber force field. The Journal of chemical physics, 153:114502, 09 2020. doi: 10.1063/5.0019056. URL https://doi.org/10.1063/5.0019056.
- Maier et al. [2015] James A. Maier, Carmenza Martinez, Koushik Kasavajhala, Lauren Wickstrom, Kevin E. Hauser, and Carlos Simmerling. ff14sb: Improving the accuracy of protein side chain and backbone parameters from ff99sb. Journal of Chemical Theory and Computation, 11(8):3696–3713, 2015. ISSN 1549-9618. doi: 10.1021/acs.jctc.5b00255. URL https://doi.org/10.1021/acs.jctc.5b00255.
- Zgarbová et al. [2011] Marie Zgarbová, Michal Otyepka, Jiří Šponer, Arnošt Mládek, Pavel Banáš, Thomas E. Cheatham III, and Petr Jurečka. Refinement of the cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. Journal of Chemical Theory and Computation, 7(9):2886–2902, 2011. ISSN 1549-9618. doi: 10.1021/ct200162x. URL https://doi.org/10.1021/ct200162x.
- Eastman et al. [2023] P. Eastman, P.K. Behara, D.L. Dotson, and et al. Spice, a dataset of drug-like molecules and peptides for training machine learning potentials. Scientific Data, 10(11), 2023. URL https://doi.org/10.1038/s41597-022-01882-6.
- Lindorff-Larsen et al. [2011] Kresten Lindorff-Larsen, Stefano Piana, Ron O. Dror, and David E. Shaw. How fast-folding proteins fold. Science, 334(6055):517–520, 2011. doi: 10.1126/science.1208351. URL https://www.science.org/doi/abs/10.1126/science.1208351.
- Rennekamp et al. [2020] Benedikt Rennekamp, Fabian Kutzki, Agnieszka Obarska-Kosinska, Christopher Zapp, and Frauke Gräter. Hybrid kinetic monte carlo/molecular dynamics simulations of bond scissions in proteins. Journal of Chemical Theory and Computation, 16(1):553–563, 2020. doi: 10.1021/acs.jctc.9b00786. URL https://doi.org/10.1021/acs.jctc.9b00786. PMID: 31738552.
- Paszke et al. [2019] Adam Paszke, Sam Gross, Francisco Massa, Adam Lerer, and et al. Pytorch: An imperative style, high-performance deep learning library, 2019.
- Wang et al. [2020] Minjie Wang, Da Zheng, Zihao Ye, Quan Gan, Mufei Li, Xiang Song, Jinjing Zhou, Chao Ma, Lingfan Yu, Yu Gai, Tianjun Xiao, Tong He, George Karypis, Jinyang Li, and Zheng Zhang. Deep graph library: A graph-centric, highly-performant package for graph neural networks, 2020.
- Clevert et al. [2016] Djork-Arné Clevert, Thomas Unterthiner, and Sepp Hochreiter. Fast and accurate deep network learning by exponential linear units (elus), 2016.
- Srivastava et al. [2014] Nitish Srivastava, Geoffrey Hinton, Alex Krizhevsky, Ilya Sutskever, and Ruslan Salakhutdinov. Dropout: A simple way to prevent neural networks from overfitting. Journal of Machine Learning Research, 15:1929–1958, 06 2014.
- Kingma and Ba [2017] Diederik P. Kingma and Jimmy Ba. Adam: A method for stochastic optimization, 2017.
- Loshchilov and Hutter [2017] Ilya Loshchilov and Frank Hutter. Sgdr: Stochastic gradient descent with warm restarts, 2017.
- Blondel and Karplus [1996] Arnaud Blondel and Martin Karplus. New formulation for derivatives of torsion angles and improper torsion angles in molecular mechanics: Elimination of singularities. Journal of Computational Chemistry, 17(9):1132–1141, 1996.
- Smith et al. [2020] D. G. A. Smith, L. A. Burns, and et al. Simmonett. Psi4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys., 2020. doi: 10.1063/5.0006002.
- [35] Berk Hess. P-LINCS: A parallel linear constraint solver for molecular simulation. 4(1):116–122. ISSN 1549-9618, 1549-9626. doi: 10.1021/ct700200b. URL https://pubs.acs.org/doi/10.1021/ct700200b.
- [36] Giovanni Bussi, Davide Donadio, and Michele Parrinello. Canonical sampling through velocity rescaling. 126(1):014101. ISSN 0021-9606, 1089-7690. doi: 10.1063/1.2408420.
- [37] M. Parrinello and A. Rahman. Polymorphic transitions in single crystals: A new molecular dynamics method. 52(12):7182–7190. ISSN 0021-8979. doi: 10.1063/1.328693. URL https://doi.org/10.1063/1.328693.
- Honda et al. [2008] Shinya Honda, Toshihiko Akiba, Yusuke S. Kato, Yoshito Sawada, Masakazu Sekijima, Miyuki Ishimura, Ayako Ooishi, Hideki Watanabe, Takayuki Odahara, and Kazuaki Harata. Crystal structure of a ten-amino acid protein. Journal of the American Chemical Society, 130(46):15327–15331, 2008. doi: 10.1021/ja8030533. URL https://doi.org/10.1021/ja8030533. PMID: 18950166.
附录A附录
A.1 格拉巴酒套餐
Grappa 在 GNU 通用公共许可证 v3.0 下作为开源软件发布,可在以下位置获取:
https://github.com/hits-mbm-dev/grappa。
它提供了一个命令行界面来重新参数化从传统力场获得的 GROMACS 拓扑文件:
grappa_gmx -f topol.top -p new_topol.top
对于 OpenMM,我们提供了一个类,可用于使用 Grappa 重新参数化系统: from grappa import OpenmmGrappa
# 使用传统的 OpenMM 力场 # 从拓扑顶部获取系统,system = …
# 下载预训练的格拉巴酒模型 ff = OpenmmGrappa.from_tag('grappa-1.1')
# 使用 grappa 重新参数化系统 system = ff.parametrize_system(system, top)
# 继续通常的工作流程
预训练发布的模型可以通过标签访问;模型权重会从 GitHub 上的相应版本自动下载。 发布的模型还包含一个包含测试数据集结果的字典、用于训练和验证的分子标识符列表以及用于在同一数据集上重现训练的配置文件。 为了复制表1和表2,可以分别使用标签“grappa-benchmark”和“grappa-1.1.0”。 有关该包的更多详细信息可以在 GitHub 上找到。
A.2模型架构
在本节中,我们将详细描述 Grappa 的架构,包括图注意力神经网络和对称 Transformer。
A.2.1 图注意力神经网络
Grappa 的图注意力神经网络(图A7)是针对图的 Transformer 架构[7]的修改,类似于图 Transformer 网络[15],但是例如省略局部性破坏图拉普拉斯特征向量作为位置编码。 相反,我们通过将注意力机制限制在图的边缘来对图结构进行编码,如 GAT [11] 中所示,这强制了局部性:在每次注意力更新中,只有相邻节点可以相互影响。 我们使用多头点积注意力机制和2层MLP作为前馈网络,其中隐藏特征维度是节点特征维度的四倍。
如下所述初始化节点特征 后,我们应用单个节点线性层,后跟指数线性单元 (ELU) [29],
| (12) |
然后,我们应用 图注意力层,每个注意力层由下式给出
| (13) | ||||
| (14) | ||||
| (15) | ||||
| (16) | ||||
| (17) | ||||
| (18) | ||||
| (19) |
其中 是节点 的邻居集合,包括节点本身, 表示注意力头,权重
| (20) | ||||
| (21) | ||||
| (22) | ||||
| (23) |
偏差 是可学习的并且对于每一层都是独立的。 我们在 (18) 和 (19) 中的节点特征更新上使用 dropout [30]。 最后,我们通过线性层投影到输出维度 上,
| (24) |
作为初始节点特征,我们选择原子序数的 one-hot 编码、节点是否在循环中的 one-hot 编码、潜在循环大小和节点度的 one-hot 编码。 由于 Grappa 仅预测键合参数,因此我们必须确保与传统力场中的非键合参数的一致性,这就是为什么如果我们在数据集上训练,我们会对用于相应状态的非键合贡献的传统力场进行 one-hot 编码包含来自不同非键合方法的数据。 我们还包括部分电荷 的正弦编码 ,如原始 Transformer 架构 [7] 中一样,
| (25) | ||||
| (26) |
与,这使我们能够描述同一分子的不同电荷构象,而无需求助于全局或图形对称性破坏特征(例如形式电荷)。
A.2.2 对称 Transformer
图注意力神经网络后面是四个(并行)对称 Transformer (图 2),一个用于每种类型的交互,即一个用于键,一个用于角度,一个用于扭转,一个用于不正确的相互作用二面体参数。 对于每个相互作用(例如分子图中的每个角度),所涉及原子的节点嵌入被映射到一组相应的 MM 参数(例如平衡角和力常数)。
如 2.2 节中所述,我们向节点特征添加对称位置编码,如 (9) 中所示,它在所需的排列集下是不变的,但可以破坏对称性这对于使模型更具表现力同时保持稍后用于不变池化 (10) 的排列下的等方差来说并不是必需的。 从对称性方程。 6-8,并根据A.4节中对不正确二面角的考虑,我们可以推导出以下位置编码
| (27) | ||||
| (28) | ||||
| (29) |
不需要键位置编码,因为键参数的排列是完整的组 并且没有需要打破的对称性。 对于扭转和不正确,位置编码不会破坏所有不必要的对称性,例如对称性仍然存在于位置编码中,并且稍后将被破坏。
然后,我们应用一个 Transformer 来充当代表参与交互的原子的一个节点,即在对维度 的特征进行投影之后,
| (30) |
我们应用 Transformer 层,每个层由具有 头的缩放点积注意力和具有隐藏维度 的 2 层 MLP 给出,跳过连接、丢失和层标准化与原始 Transformer 架构[7] 中一样。
然后,我们通过将串联节点嵌入的排列版本传递到具有隐藏维度 的 层 MLP 并对所有排列 求和来应用对称池操作各自的对称群,
| (31) |
键的二维分数 和角度的 最终通过 ELU 和 sigmoid 函数的缩放和移位版本映射到相应的 MM 参数集的范围,
| (32) | ||||
| (33) | ||||
| (34) | ||||
| (35) |
其中ToPos和在A.2.3节中定义,表示第得分向量的条目。 对于二面体力常数, 维度分数 通过 sigmoid 门馈送,以使模型对于小力常数更具表现力,
| (36) |
A.2.3 神经网络输出的缩放
为了将我们的模型预测的分数映射到相应 MM 参数的范围,我们使用 sigmoid 函数的缩放和移位版本来映射到指定的区间,并使用 ELU 来映射到正值。 我们选择这些函数是因为它们是可微的,并且与指数函数相反,它们仅在大输入时线性增长,这有助于稳定训练。 为了使模型能够预测顺序统一或近似正态分布的分数,我们选择缩放和移位参数,使得正态分布的输入将导致输出分布的均值和标准差接近给定值目标平均值和标准差。 我们通过计算训练数据给定子集上传统 MM 力场参数的平均值和标准差来选择目标标准差(平衡角除外,我们将 限制为 ) 。
为了映射到正值,我们使用
| (37) |
为了映射到指定的间隔 ,我们将自己限制在 使用的情况
| (38) |
如果我们考虑 sigmoid 函数和 ELU 在输入接近零时的渐近行为,我们可以看到输出的均值和标准差确实接近目标均值和标准差,
| (39) | ||||
| (40) |
这是由 ELU 和 sigmoid 函数在零附近的泰勒展开得出的。 因此,利用 具有消失的均值和单位标准差,我们确实可以对 进行一阶排序,
和
为了映射到正值,我们将 中的 ELU 扩展到零附近,如果 则这是合理的,并类比地发现目标均值和标准差在 。
A.3 训练超参数和详细信息
| Hyperparameter | Variable | Value |
| Graph Neural Network | ||
| Layers | 7 | |
| Hidden dimension | 512 | |
| Attention heads | 16 | |
| Embedding dimension | 256 | |
| Dropout probability | 0.3 | |
| Symmetric Transformer | ||
| Transformer layers | 3 | |
| Transformer hidden dim | 512 | |
| Attention heads | 8 | |
| Pooling layers | 3 | |
| Pooling hidden dim | 256 | |
| Dropout probability | 0.5 | |
| Training setup | ||
| Learning rate | ||
| Molecules per batch | 32 | |
| States per molecule | 32 | |
| Force weight | 0.8 | |
| Dihedral weight | ||
| Traditional MM weight | 0 | |
对于本工作中讨论的所有模型,我们使用表 A3 中列出的超参数和 的二面周期。 能量、力和 MM 参数之间的所有相对权重都依赖于 kcal/mol、Å 和弧度形成的单位。 我们使用 Adam [31] 优化器和 、、 来训练模型。
首先,我们仅在传统 MM 参数上训练 2 个 epoch,这可以解释为预训练的一种形式,它可以有效地使模型处于损失函数 QM 部分的梯度提供信息的状态。 我们从 开始能量和力的训练,在 100 个周期后将其设置为零,以防止模型收敛到损失函数 QM 部分的非物理局部最小值。 当如上所述改变损失函数项的权重时,我们用随机时刻重新初始化优化器并应用热重启[32],在此期间我们以 500 个训练步骤线性增加学习率将优化器状态与先前损失函数的梯度更新去相关。 除了直接对传统MM参数进行预训练外,我们还发现惩罚大扭转和不适当的力常数有利于跨构象空间的泛化性。
A.4 不正确的二面角
正如 2.2 节中所讨论的,我们假设不同相互作用的能量贡献在所涉及原子的嵌入和空间位置的某些排列下是对称的。 这些排列由描述相互作用的各个子图的同构给出。 在角度、键和扭转的情况下,这些对称性由方程式给出。 6-8,对于不等边二面体,对称性由所有六种使中心原子不变的排列给出,这可以从图 A8 中子图的结构看出。 对于键、角度和扭转,相互作用坐标距离、角度和二面角的余弦在各自的排列下是不变的,这就是为什么我们可以简单地通过在各自的排列下强制MM参数的不变性来构造不变的能量贡献。
对于不正确的二面角,情况并非如此,因为二面角在上述所有六种排列下都不是不变的,并且具有在这些排列下不变的不正确力常数的模型不会具有不变的不正确能量贡献。 在 Grappa 中,我们通过引入更多不正确二面角的术语来解决这个问题。 例如
| (41) |
在排列 下是不变的,可以推广到所有六种不正确的排列。 事实证明,我们可以通过使用二面角的另一种对称性将附加项的数量减少到两项,
| (42) |
从[33]中的二面角公式可以看出。 这种对称性使我们能够识别在排列 下相互转换的项对,并使用在此排列下不变的力常数,
| (43) |
A.5数据集
A.5.1 Espaloma数据集
通过下载已发布的预处理数据222https://github.com/hits-mbm-dev/grappa/blob/master/dataset_creation/get_espaloma_split/load_esp_ds.py,我们重现了用于训练和评估 Espaloma 0.3 的数据集[10]。 预处理需要过滤掉高能构象,如[10]中详述。 与 Espaloma 一样,我们的训练-验证-测试分区程序依赖于分子的独特异构 SMILES 字符串,这些字符串包含在数据集中。 我们重现333https://github.com/hits-mbm-dev/grappa/blob/master/dataset_creation/get_espaloma_split/load_esp_ds.py Espaloma 0.3444https://github.com/choderalab/refit-espaloma/blob/main/openff-default/02-train/joint-improper-charge/charge-weight-1.0/train.py, commit 3ccc44d。
A.5.2 数据集创建
用于创建数据集的脚本可以在以下位置找到 https://github.com/LeifSeute/grappa-data-creation。 我们使用 Amber ff99SB-ILDN [17] 力场对状态进行采样,并使用 Psi4 [34] 进行单点 QM 计算。 为了对自由基肽状态进行采样,我们使用了经过优化轨迹和自由基肽扭转扫描训练的 BIKGrappa 模型。 自由基肽就是我们所说的氢原子转移(HAT)型自由基,即它们是通过从肽中除去单个氢而形成的。 对于非键合贡献,我们使用与原始肽相同的 Lennard-Jones 参数,并将原始肽中氢的部分电荷添加到它所附着的重原子上。
A.6MD模拟
所有折叠和稳定性模拟均使用 GROMACS 版本 2023 [8] 使用 Amber ff99SB-ILDN 力场 [17] 或使用 Amber ff99SB-ILDN 非键合参数的 Grappa-1.1 进行和TIP3P水模型。 采用 2 fs 时间步长,对氢键采用 LINCS 约束[35]。 模拟在 300K 和 1 bar 下运行,由 v-rescale 恒温器 [36] 和 Parrinello-Rahman 压力耦合 [37] 维持。 我们应用 1.0ṅm 的库仑和伦纳德-琼斯截止值。 系统准备完毕后,进行能量最小化、NVT 和 NPT 平衡模拟。
用于重现模拟的代码可以在以下位置找到
https://github.com/LeifSeute/validate-grappa。
A.6.1 折叠模拟
对于折叠模拟,我们遵循 Lindorff-Larsen 等人的模拟系统设置。 [25] 从完全扩展的溶剂化结构中,我们最小化并平衡,然后进行短 MD 模拟,其中蛋白质的端到端距离通常会缩短。 我们选择一个端到端距离较小的状态,并将其作为实际折叠模拟的起点,其中我们可以选择较小的模拟框,从而降低计算成本。 对于具有序列 YYDPETGTWY 的 chignolin [38] 突变体(PDB ID:5AWL)的折叠模拟,我们选择 40 Å 的盒子大小,并用两个钠离子中和系统。
A.6.2 稳定性模拟
通过计算初始结构的 C-alpha RMSD 0.05 ns 内的移动平均值来评估泛素(PDB ID:1UBQ)的模拟(图4b)。 我们还计算了 C-alpha RMSD 的统计数据作为两个状态之间时间差的函数。 为此,我们从每个时间差的轨迹中采样 1,000 个状态,并计算状态之间的平均 C-alpha RMSD。
| Dataset | Test Mols | Confs | Grappa | Espaloma | Gaff-2.11 | ff14SB, | Mean | |
| RNA.OL3 | Predictor | |||||||
| BOLTZMANN SAMPLED | ||||||||
| SPICE-Pubchem | 1411 | 60853 | Energy | 2.3 0.1 | 2.3 0.1 | 4.6 | 18.4 | |
| Force | 6.1 0.3 | 6.8 0.1 | 14.6 | 23.4 | ||||
| SPICE-DES-Monomers | 39 | 2032 | Energy | 1.3 0.1 | 1.4 0.3 | 2.5 | 8.2 | |
| Force | 5.2 0.2 | 5.9 0.5 | 11.1 | 21.3 | ||||
| SPICE-Dipeptide | 67 | 2592 | Energy | 2.3 0.1 | 3.1 0.1 | 4.5 | 4.6 | 18.7 |
| Force | 5.4 0.1 | 7.8 0.2 | 12.9 | 12.1 | 21.6 | |||
| RNA-Diverse | 6 | 357 | Energy | 3.3 0.2 | 4.2 0.3 | 6.5 | 6.0 | 5.4 |
| Force | 3.7 0.04 | 4.4 0.1 | 16.7 | 19.4 | 17.1 | |||
| RNA-Trinucleotide | 64 | 35811 | Energy | 3.5 0.1 | 3.8 0.2 | 5.9 | 6.1 | 5.3 |
| Force | 3.6 0.01 | 4.3 0.1 | 17.1 | 19.7 | 17.7 | |||
| TORSION SCAN | ||||||||
| Gen2-Torsion | 131 | 21890 | Energy | 1.7 0.2 | 1.6 0.3 | 2.7 | 4.7 | |
| Force | 4.0 0.4 | 4.7 0.6 | 9.4 | 5.5 | ||||
| Protein-Torsion | 9 | 6624 | Energy | 2.2 0.4 | 1.9 0.2 | 3.0 | 3.5 | |
| Force | 3.8 0.5 | 3.5 0.3 | 9.7 | 5.1 | ||||
| OPTIMIZATION | ||||||||
| Gen2-Opt | 154 | 40055 | Energy | 1.8 0.2 | 1.7 0.5 | 3.0 | 3.9 | |
| Force | 3.8 0.2 | 4.5 0.8 | 9.7 | 5.1 | ||||
| Pepconf-Opt | 55 | 14884 | Energy | 3.2 0.3 | 2.8 0.3 | 5.1 | 4.1 | 6.3 |
| Force | 3.6 0.2 | 4.0 0.4 | 10.2 | 10.2 | 5.3 | |||
| Dataset | Test Mols | Confs | Grappa | Grappa | Espaloma | ff14SB, | Mean | |
| AM1-BCC | ff99SB | RNA.OL3 | Predictor | |||||
| BOLTZMANN SAMPLED | ||||||||
| SPICE-Pubchem | 1411 | 60853 | Energy | 2.3 | 2.3 | 18.6 | ||
| Force | 6.1 | 6.8 | 23.5 | |||||
| SPICE-DES-Monomers | 39 | 2032 | Energy | 1.3 | 1.4 | 8.3 | ||
| Force | 5.3 | 5.9 | 21.3 | |||||
| SPICE-Dipeptide | 67 | 2592 | Energy | 2.3 | 2.2 | 3.1 | 4.6 | 4.3 |
| Force | 5.5 | 5.9 | 7.8 | 12.1 | 10.7 | |||
| RNA-Diverse | 6 | 357 | Energy | 3.4 | 4.2 | 6.0 | 5.8 | |
| Force | 3.7 | 4.4 | 19.4 | 17.0 | ||||
| RNA-Trinucleotide | 64 | 35811 | Energy | 3.6 | 3.8 | 6.1 | 5.8 | |
| Force | 3.6 | 4.3 | 19.7 | 17.8 | ||||
| Radical-Dipeptide | 28 | 275 | Energy | 2.8 | 7.4 | |||
| Force | 6.5 | 23.6 | ||||||
| HYP-DOP-Dipeptide | 8 | 363 | Energy | 2.3 | 6.8 | |||
| Force | 5.6 | 24.9 | ||||||
| Uncapped-Dipeptide | 12 | 600 | Energy | 2.1 | 3.7 | 6.5 | ||
| Force | 5.9 | 11.7 | 25.3 | |||||
| TORSION SCAN | ||||||||
| Gen2-Torsion | 131 | 21890 | Energy | 1.8 | 1.6 | 4.8 | ||
| Force | 4.0 | 4.7 | 5.5 | |||||
| Protein-Torsion | 9 | 6624 | Energy | 2.2 | 2.2 | 1.9 | 2.4 | |
| Force | 3.9 | 5.2 | 3.5 | 9.6 | ||||
| OPTIMIZATION | ||||||||
| Gen2-Opt | 154 | 40055 | Energy | 1.8 | 1.7 | 3.9 | ||
| Force | 3.8 | 4.5 | 5.1 | |||||
| Pepconf-Opt | 55 | 14884 | Energy | 3.2 | 2.7 | 2.8 | 4.1 | 3.5 |
| Force | 3.7 | 4.5 | 4.0 | 10.2 | 9.6 | |||