ProteinBench:蛋白质基础模型的整体评估
摘要
近年来,蛋白质基础模型的开发蓬勃发展,显著提高了蛋白质预测和生成任务的性能,从 3D 结构预测和蛋白质设计到构象动力学。 然而,由于缺乏统一的评估框架,这些模型的能力和局限性仍然知之甚少。 为了填补这一空白,我们引入了 ProteinBench,这是一个旨在提高蛋白质基础模型透明度的整体评估框架。 我们的方法包括三个关键组成部分:(i)基于不同蛋白质模式之间关系的蛋白质领域主要挑战的分类法分类;(ii)一种多指标评估方法,评估四个关键维度的性能:质量、新颖性、多样性和鲁棒性;以及(iii)来自各种用户目标的深入分析,提供模型性能的整体视图。 我们对蛋白质基础模型的全面评估揭示了一些关键发现,这些发现揭示了它们当前的能力和局限性。 为了促进透明度并促进进一步的研究,我们发布了评估数据集、代码和公开排行榜,以供进一步分析和通用模块化工具包。 我们希望 ProteinBench 成为一个活生生的基准,为建立蛋白质基础模型的标准化、深入评估框架奠定基础,推动其发展和应用,同时促进该领域内的合作。
1简介
蛋白质是基本分子,在各种生物过程中发挥着关键作用,从酶催化和信号转导到结构支持和免疫反应。 它们的功能由其氨基酸序列决定,通常通过折叠成特定的三维结构来介导。 了解蛋白质序列、结构和功能之间复杂的相互作用对于推进涵盖制药、农业、特种化学品和生物燃料的科学和工程至关重要 (Kuhlman & Bradley, 2019).
近年来,蛋白质基础模型的开发呈井喷式发展 1 11在本研究中,我们将蛋白质基础模型的定义扩展到包括任何旨在解决蛋白质科学基础问题的生成模型。 旨在通过捕捉蛋白质的复杂机制来理解基本的生物过程 (Jumper et al., 2021; Abramson et al., 2024; Lin et al., 2023; Watson et al., 2023b; Ingraham et al., 2023; Krishna et al., 2024; Shin et al., 2021; Madani et al., 2023; Alley et al., 2019; Wang et al., 2024b; Hayes et al., 2024; Hie et al., 2024). 这些模型利用先进的深度学习和生成式人工智能技术,展示了非凡的能力,标志着从传统的、特定于任务的方法向更具泛化能力的框架的重大转变,这些框架能够学习庞大蛋白质数据集中的复杂模式和关系。 例如,AlphaFold3 (Abramson et al., 2024) 基于扩散模型,在所有生物分子的全原子结构预测方面取得了前所未有的精度,而其他模型,如 ESM 系列 (Rives et al., 2021; Hsu et al., 2022; Lin et al., 2023; Verkuil et al., 2022; Hayes et al., 2024) 和 DPLM (Wang et al., 2024b) 在蛋白质语言建模方面表现出令人印象深刻的表示能力,有利于各种下游任务。 此外,这些基础模型并不局限于单一模态。 同时考虑序列、结构和功能的多模态模型正在出现,为全面理解蛋白质行为提供了可能 (Hayes et al., 2024; Liu et al., 2023)。 理解这种序列-结构-功能关系的一个重要方面是蛋白质构象动力学。 最近的工作将蛋白质结构预测扩展到几个构象预测任务,并将生成式人工智能引入到蛋白质构象分布的模型中 (Jing et al., 2023; Zheng et al., 2024; Jing et al., 2024; Wang et al., 2024c; Lu et al., 2024)。
然而,蛋白质基础模型的快速发展也迫切需要一个统一的框架来全面评估其在各种任务、数据集和指标上的性能,如附录 A 所示。 当前的蛋白质基础模型格局的特点是不统一的建模方法、特定于任务或特定于模型的评估标准。 评估方法的这种异质性使得在不同模型之间进行有意义的比较以及全面了解其相对优势和局限性变得具有挑战性。
通过对跨越不同生物领域的各种数据集进行系统评估,特别是在蛋白质设计和构象动力学方面,我们的目标是提供对蛋白质基础模型的模型架构和性能的全面分析。 这种方法使我们能够剖析各种模型组件和数据特征对蛋白质建模不同方面的影响。 在标准化基准测试中比较这些模型的能力对于指导未来的研究方向、为实际应用提供模型选择信息以及推动整个领域的发展至关重要。
在本研究中,如图 1 所示,我们介绍了 ProteinBench,这是第一个旨在通过四个关键组件对蛋白质基础模型进行全面评估的基准测试:
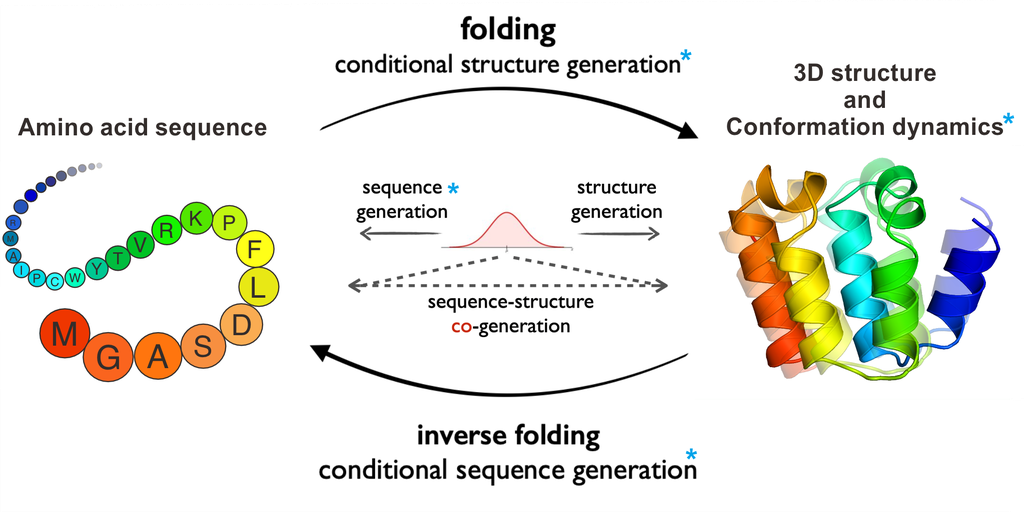
(1) 包含蛋白质领域主要生成挑战的任务分类。 ProteinBench涵盖了广泛的生成任务,包括蛋白质设计(涵盖结构设计、序列设计、结构-序列协同设计以及抗体设计的特定应用任务)、三维结构预测和构象动力学。 这些任务针对不同的蛋白质模态,能够对模型架构和模态特征对性能相互作用进行细致的分析。 我们利用多样化且精心策划的数据集来捕捉蛋白质宇宙的复杂性和多样性,确保对模型能力进行全面评估。
(2) 一种多指标评估方法,评估四个关键维度的性能:质量、新颖性、多样性和鲁棒性。 目前对蛋白质生成模型的评估通常存在非统一的指标和不完整的评估,通常只关注一两个方面。 然而,蛋白质科学问题包含一组复杂而系统的挑战。 蛋白质建模和设计中的下游任务涉及序列、结构和功能之间的复杂相互作用。 ProteinBench通过提供对模型捕获蛋白质宇宙机制的能力的全面衡量来解决此限制。 我们根据四个关键维度评估模型:质量、新颖性、多样性和鲁棒性。 这种多方面的做法提供了对模型性能和能力的更全面视角。
(3) 来自不同用户目标的深入分析,提供了模型性能的整体视图。 认识到不同的用户在应用蛋白质基础模型时可能会有不同的目标,我们从多个角度进行了深入分析。 例如,在蛋白质设计中,一些用户可能优先考虑符合自然进化分布的模型,而另一些用户可能寻求能够生成训练集分布之外的新型蛋白质的模型。 通过从这些不同的目标分析模型能力,ProteinBench提供了对广泛的实际应用有益的见解。
(4) 排行榜和代码框架。 为了便于公平比较并支持新方法的开发,我们提供了一个统一的实验框架。 这包括一个公共排行榜和开源代码,使研究人员能够轻松地将他们的模型与现有模型进行基准测试,并为该领域的持续发展做出贡献。
通过整合这四个组件,ProteinBench旨在为蛋白质基础模型建立一个标准化、全面且以用户为中心的评估框架。 这种方法不仅揭示了当前的最佳技术水平,而且指引了未来的研究方向,并加速了蛋白质建模和设计领域的进展。
2 背景和任务定义
在本节中,我们将简要概述各种蛋白质基础模型所解决的任务,如表 1所示,重点关注两个关键的生成任务:蛋白质设计和构象动力学。 这两个领域进一步细分为八个子任务。
| Tasks | Dimension | Metrics | Methods |
|---|---|---|---|
| Protein Design | |||
| Inverse Folding | Sequence recovery | AAR | ProteinMPNN, ESMIF1, |
| Refoldability | scTM (AF2) | LM-Design, ESM3 | |
| Stability | pLDDT (AF2) | PiFold, CarbonDesign | |
| Backbone Design | Quality | scTM, scRMSD (ProteinMPNN & ESMFold) | Rfdiffusion, Frameflow, Chroma, |
| Novelty | Max. TM score to PDB database (Foldseek) | Framediff, Foldflow, Genie | |
| Diversity | Pairwise TM, Max Cluster (Foldseek) | foldingdiff, Proteus | |
| Sequence Design | Quality | pLDDT (AF2) | ProGen2, EvoDiff, |
| Novelty | Max. TM to PDB database (Foldseek) | DPLM, ESM3 | |
| Diversity | Pairwise TM , Max Cluster (Foldseek) | ||
| Struct-seq Co-design | Quality | scTM, scRMSD (ESMFold) | ProteinGenerator, ProtPardelle, |
| Novelty | Max. TM score to PDB database (Foldseek) | Multiflow, ESM3, CarbonNovo | |
| Diversity | Pairwise TM, Max Cluster (Foldseek) | ||
| Motif Scaffolding | Quality | Motif RMSD, Scafold RMSD | FrameFlow, Rfdiffusion, TDS, EvoDiff, DPLM, ESM3 |
| Antibody Design | Accuracy | AAR, RMSD, TM-score | HERN, |
| Functionality | Binding Energy (Rosetta) | MEAN, dyMEAN, | |
| Specificity | Seq Similarity, PHR | DiffAb, AbDPO | |
| Rationality | CN-Score, Clashes, Seq Naturalness | ||
| Total Energy (Rosetta), scRMSD (IgFold) | |||
| Protein Conformation Prediction | |||
| Single state (folding) | Accuracy | TM score, RMSD, GDT, lDDT | AlphaFold2, OpenFold, ESMFold, |
| Quality | CA clash/break rate, Peptide bond break rate | RosettaFold2, EigenFold | |
| Multiple state Prediction | Accuracy | Ensemble TM score/RMSD | EigenFold, MSA-subsampling, Str2Str, AlphaFlow/ESMFlow, ConfDiff |
| Diversity | pairwise RMSD/TM | ||
| Quality | CA clash/break rate, Peptide bond break rate | ||
| Distribution Prediction | Accuracy | Flexibility accuracy, Distributional similarity, Ensemble observables | |
| Diversity | Pairwise RMSD, RMSF | ||
| Quality | CA clash/break rate, Peptide bond break rate | ||
对于每个任务,我们关注以下方面,附录中提供了详细信息:
[任务定义] 清晰简洁地描述任务,包括其目标和与蛋白质科学的相关性。 每个任务的输入数据格式和预期输出的规范。
[评估指标] 用于评估模型性能的指标描述,包括质量、新颖性、多样性和鲁棒性度量。
[数据集] 用于每个任务的数据集概述,包括它们的大小、多样性和任何应用的预处理步骤。
2.1 蛋白质设计
2.1.1 逆折叠
[任务定义] 目标是预测给定目标蛋白质结构的最佳氨基酸序列,同时考虑稳定性、可重折叠性和潜在功能等因素。
[评估指标] 蛋白质序列设计的性能使用多个互补指标进行评估:(1) 序列恢复: 此指标将设计的序列与具有类似结构的天然序列进行比较。 它量化了设计方法在多大程度上能够概括与特定结构基序相关的进化保守序列模式。 (2) 可重折叠性: 此指标评估目标主干与设计序列预测结构之间的结构相似性。 使用 AlphaFold2 (Jumper 等人,2021) 进行预测。 使用自洽模板建模评分 (scTM) (Trippe 等人,2022) 和自洽均方根偏差 (scRMSD) 来量化相似性,从而深入了解设计序列在多大程度上能够折叠成预期结构。 (3) 稳定性: 使用 AlphaFold2 计算的预测局部距离差异测试 (pLDDT) 来评估。 pLDDT 分数用作设计蛋白质预测稳定性的代理指标,用于 Dauparas 等人 (2022)。
[数据集] 评估是在针对结构基础序列设计的两个不同目标的不同数据集上进行的:(1) 捕获本地进化分布: 我们评估了包含新发布的 PDB 结构的两个独立数据集:CASP15 (cas, 2022) 和 CAMEO (Robin 等人,2021)。 我们从 2024 年 1 月到 7 月的持续 CAMEO 评估中收集了新结构,总共 332 个复杂结构。 此外,从 CASP15 收集了 32 个蛋白质结构,其中仅包括蛋白质实体,不包括核酸或配体。 (2) 从头蛋白质设计: RFdiffusion (Watson 等人,2023a) 用于生成不同长度的主干:具体来说,100、200、300、400 和 500 个残基。 对于每种长度,随机采样 10 个不同的结构,所有方法的采样温度为 0.1。 使用 AlphaFold2 评估这些序列的可设计性,其中 scTM 分数和 pLDDT 指标作为主要评估标准。 现有的逆折叠基准测试,例如 PDB-Struct (Wang 等人,2023) 和 Proteininvbench (Gao 等人,2024),为评估逆折叠方法提供了标准化的蛋白质结构集。 尽管这些基准测试已为该领域的进步做出了重大贡献,但对更全面的评估框架的需求日益增长。 这些扩展的评估应该更贴近蛋白质设计中不同的用户目标,包括捕捉自然进化分布的准确性和从头开始基于骨架的序列设计的鲁棒性等方面。
2.1.2 蛋白质骨架设计
[任务定义] 蛋白质骨架设计侧重于创建新的蛋白质折叠以实现从头开始的设计目标。 这项任务对于扩展蛋白质结构的范围(超出自然界中发现的范围)至关重要,在药物发现、生物材料和治疗等领域具有重要应用。
[评估指标] 骨架设计的评估涵盖多个标准,以评估生成的结构的质量和新颖性。 结构 质量 主要使用自洽 TM-score 和 RMSD 测量,它们提供骨架可折叠性的定量指标,通过 ProteinMPNN (Dauparas 等人,2022) 和 ESMFold (Lin 等人,2023) 测量。 同样重要的是 新颖性 指标,它们衡量方法探索超出已知蛋白质折叠的新结构空间的能力。 这方面使用两个关键指标进行评估:在将设计的结构与 RCSB 蛋白质数据库 (PDB) (Berman 等人,2000) 中的现有条目进行比较时获得的最大 TM-score。 此比较是使用快速结构对齐工具 Foldseek (van Kempen 等人, 2022) 进行的。 多样性 指标,包括:(a) 设计结构之间的成对最大 TM-score。 (b) 在设计骨架集中识别的不同结构簇的数量,也使用 Foldseek (van Kempen 等人,2022) 确定。 这些多样性指标有助于量化设计方法可以产生的独特结构范围,确保它不仅仅是在重新创建已知的折叠,而是在生成各种蛋白质骨架库。
[数据集] 生成任务的主要目标是准确地映射训练集的一般分布。 对于蛋白质结构生成,通常使用来自蛋白质数据库 (PDB) 的高分辨率结构。 为了深入了解这种数据分布,我们从RCSB数据库中随机抽取了100个天然单链结构作为参考。 为了确保多样性,我们迭代地移除与其他结构相比具有最高TM分数的结构,直到我们得到最终的100个不同结构集。 这种方法提供了PDB中单链结构分布的代表性快照,作为评估生成模型在捕捉蛋白质结构真实分布方面的性能的基准。
2.1.3 蛋白质序列设计
[任务定义] 该任务的目的是生成具有所需性质的氨基酸序列,例如质量、多样性和新颖性。
除了基于序列的评估,生成的序列的结构特征也很重要。
[评估指标] 对于序列自然度,我们使用来自自回归蛋白质语言模型 (ProGen2) 的困惑度来量化生成的序列模式是否位于自然序列分布中。
对于基于结构的评估,我们使用单序列折叠模型,即ESMFold,来预测生成序列的结构,然后使用pLDDT作为序列的结构稳定性的代理,通过AlphaFold2预测的结构来测量结构质量,以及使用与主链设计中相同的协议来测量结构多样性和新颖性。
[数据集] UniRef50是训练蛋白质序列生成模型和语言模型常用的数据集。
2.1.4 结构和序列协同设计
[任务定义] 蛋白质结构-序列协同设计涉及同时优化蛋白质的主链结构和氨基酸序列,以实现所需的性质或功能。 与单独的序列设计或结构设计相比,此任务更加复杂,因为它探索了更大的解决方案空间。
[评估指标] 评估指标源自用于序列和结构设计的指标:结构质量评估、序列-结构兼容性,以及序列和结构相对于已知蛋白质的新颖性也至关重要。
[数据集] 来自蛋白质数据库 (PDB) 的高分辨率蛋白质结构是此任务常用的数据集,同时要仔细考虑以消除冗余。
2.1.5 基序支架
[任务定义] 基序支架涉及设计一个包含特定功能基序或结合位点的蛋白质结构。 目标是创建一个稳定的蛋白质框架(支架),以正确的几何形状呈现所需的基序,以发挥其功能。
[评估指标] 遵循 Yim 等人(2024),关键指标包括设计支架中基序的结构准确性(通常通过 RMSD 测量)、整体蛋白质稳定性和基序功能特性的保留。 通过结合测定或酶活性测试进行的实验验证通常至关重要。
[数据集] 数据集通常包括已知功能基序库(例如,催化位点、结合界面)和可以潜在地容纳这些基序的不同支架结构。
蛋白质数据库是主要来源,但功能位点的策划数据集,例如催化位点图谱,也很有价值。
[相关基准] 酶设计挑战提供了相关的测试案例。 但是,鉴于基序支架任务的特殊性,基准通常需要针对正在目标化的特定基序或功能类别进行定制。 目前,该领域还没有针对这项任务的综合基准。
RFDiffusion 中使用了一个广泛使用的基准,其中包含 17(25)个基序支架问题 (Watson 等人,2023b)。
2.1.6 抗体设计
[任务定义] 抗体设计的目标是产生能够特异性结合给定抗原的抗体。 由于抗体的互补决定区 (CDR) 变化很大,并且主要负责抗原结合,因此抗体设计可以简化为 CDR 区域的设计,并进一步简化为重链中第三个 CDR (CDR-H3) 的设计。 鉴于蛋白质结构在相互作用中起着至关重要的作用,抗体设计通常涉及在结合抗原时同时设计序列和结构。
[评估指标] 作为一项高度面向目标的功能性蛋白质设计任务,抗体设计的评估很简单,即 功能性(与目标抗原的结合能力)和 特异性 的设计抗体。 此外,需要评估设计抗体序列和结构的 合理性 以过滤掉无效的设计。 现有研究还通过测量设计抗体与天然抗体的相似性来评估设计抗体的 准确性,因为已确认天然抗体是有效的。 然而,在许多情况下,使用准确性作为评估指标是不充分的,我们将在第3.1.6节中详细说明。
[数据集] 结构抗体数据库 (SAbDab Dunbar 等人 (2013)) 是抗体设计中常用的数据集。 它包含抗体-抗原复合物的结构数据,但数据量有限且包含大量冗余。
2.2 蛋白质构象预测
2.2.1 蛋白质折叠:单态预测
[任务定义] 蛋白质折叠的任务是根据蛋白质的序列预测其折叠结构。 折叠模型,如 AlphaFold2,在最近的蛋白质构象预测模型开发中发挥了关键作用 (Jing 等人,2024; Wang 等人,2024c)。 因此,我们认识到将蛋白质折叠纳入此基准的必要性,将其视为单一构象状态的蛋白质构象预测的具体实例。
[评估指标] 使用 RMSD、TM-score、全局距离测试 (GDT) 和局部距离差异测试 (lDDT) 将预测结构与存储在 PDB 中的参考结构进行比较,以评估预测结构的准确性。 我们还通过测量预测结构中碰撞的 α 碳原子 (CA-clash)、断开的相邻 α 碳原子 (CA-break) 和断开的肽键 (PepBond-break) 的比率来评估预测结构的质量。 详细信息请参见附录B.2.2。
[数据集] 本基准中比较的大多数折叠模型是在 2022 年之前建立的。 我们使用来自Jing 等人 (2023) 的 CAMEO2022 进行评估,该数据集包含 183 条短至中等长度的单蛋白质链 ( 750 个氨基酸),来自 2022 年 8 月 1 日至 10 月 31 日之间的 CAMEO 目标。
2.2.2 多态预测
[任务定义] 作为单态预测任务的扩展,多态预测旨在通过采样准确地预测蛋白质在不同条件下(例如配体结合)或通过分子动力学模拟观察到的两个或多个不同的构象状态。 除折叠结构外,预测这些“替代”构象的能力可以提供对构象变化和蛋白质功能的见解。
[评估指标] 我们根据准确性、多样性和质量来评估这项任务。 预测状态的准确性 由样本与参考结构的最佳结构相似性决定,通过 TM-score 或 RMSD 衡量。 多状态预测的总体准确性通过“集成准确性”评估,即所有参考状态的平均准确性(TMens 或 RMSDens,其中“ens”代表集成),类似于Jing 等人 (2023)。 对于样本结构多样性,我们测量样本之间的成对 TM-score(或 RMSD)。 最后,我们评估生成的样本的结构质量,类似于单状态预测,使用 CA-clash、CA-break 和 PepBond-break。
[数据集]
我们在两个来自先前工作的公开数据集上对模型进行基准测试:1)apo-holo,其中包含 91 种蛋白质,每种蛋白质都有一对实验结构(apo 或未结合,以及 holo 或结合),与配体结合诱导的构象变化相关 (Saldaño 等人,2022;Jing 等人,2023);(2) BPTI(牛胰蛋白酶抑制剂),一种含有 58 个氨基酸的蛋白质,先前的一项长时间 MD 模拟揭示了五种不同的构象簇 (Shaw 等人,2010)。
2.2.3 分布预测
[任务定义] 与多状态预测形成对比,多状态预测的主要目标是恢复特定的构象状态,而分布预测则侧重于生成类似于目标分布的样本分布——例如从分子动力学(MD)采样的经验分布。 这一任务进一步弥合了蛋白质构象预测模型与目前用于研究蛋白质动力学和热力学性质的基于 MD 的方法之间的差距。
[评估指标] 除了之前部分的质量 和 多样性 标准外,我们遵循(Jing 等人,2024),并包括三类指标来比较模型生成的样本集合与来自 MD 模拟的参考样本:灵活性 评估模型是否可以区分更“灵活”的区域或蛋白质与更“不灵活”的区域或蛋白质,通过区域/蛋白质多样性的 Pearson 相关性 衡量(例如,成对 RMSD);分布准确性 通过 Wasserstein 距离或第一个主成分的余弦相似性,直接比较模型生成的样本的构象分布与参考 MD 构象;以及 集成可观察量 聚焦于与功能相关的可观察量,例如由于动力学而导致的残基之间的瞬时接触,并将样本集合与来自 MD 的参考集合进行比较。 有关指标的详细说明,请参见附录 B.2.2。
[数据集] 我们使用 ATLAS 数据集 (Vander Meersche 等人,2024) 评估性能,这是一个最近发布的用于各种蛋白质的 MD 模拟结果数据库。 为了避免对在 ATLAS 数据集的部分内容上训练的模型进行数据泄露,我们遵循Jing 等人 (2024),并在 82 种蛋白质上进行基准测试,这些蛋白质的 PDB 条目是在 2019 年 5 月 1 日之后提交的,并且不属于训练集或验证集的一部分。
3 ProteinBench
在本节中,我们提供 ProteinBench,这是一个用于蛋白质基础模型的整体评估框架。 通过系统地评估蛋白质基础模型在以下任务上的表现,我们旨在全面了解其能力和局限性。 这种方法允许对不同的模型架构和策略进行细致的比较,突出优势领域,并确定改进的机会。 该基准测试中使用的所有数据都可公开获取,确保可重复性和促进研究界更广泛的参与。
3.1 蛋白质设计
在本节中,我们对各种蛋白质基础模型在基本蛋白质设计任务上的表现进行了全面的评估,包括单模态方法(基于结构的序列设计、结构设计和序列设计)、多模态结构-序列协同设计以及抗体设计的特定应用任务。 这种全面的评估使我们能够考察不同建模方法在各种蛋白质工程挑战中的多功能性和有效性。 值得注意的是,对于主链设计、序列设计、协同设计和基序支架,质量、新颖性和多样性指标使用相同的方法计算。 通过在任务中使用共同的评估指标,我们能够进行跨任务比较,希望提供性能分析以识别每种建模方法的优势和局限性,并帮助揭示不同蛋白质模态之间的潜在协同作用,以便进行未来的研究。
3.1.1 逆折叠
在本节中,我们评估了各种逆折叠模型在基于结构的序列设计方面的性能,重点关注两个不同的目标:自然进化适应度(分布内蛋白质)和从头设计的基于主链的序列设计。 后者代表一个分布外问题,测试了方法的稳健性,因为这些结构通常包含一些与 PDB 中沉积的高分辨率结构不同的噪声。 结果如表 2 所示。
我们对本地分布适应度的分析表明,基于语言模型的方法,例如我们在研究中的 LM-Design (Zheng et al., 2023),有效地捕捉到了自然进化分布,在基于本地蛋白质结构的序列设计中实现了很高的序列恢复率。 这表明这些模型有效地学习并复制了通过进化过程而产生的氨基酸选择错综复杂的模式。 然而,当应用于从头设计的基于主链的序列设计时,它的性能会下降。 相反,ProteinMPNN (Dauparas et al., 2022),一种专门为从头设计而开发并使用添加了 0.2Å 噪声的扰动坐标进行训练的方法,在从头设计任务中始终表现出优异的性能。 然而,当在适应本地进化的目标上评估时,ProteinMPNN 的性能会下降。 该发现对该领域具有重大意义,表明目前没有单一模型在所有蛋白质设计目标中都能胜出。 模型的选择应与预期应用仔细匹配。
ESM-IF1 (Hsu 等人,2022) 在基于 GVP (Jing 等人,2020) 和 Transformer 架构的 AlphaFoldDB (Varadi 等人,2022) 中最大的原生序列和结构数据集上进行训练,并在训练过程中加入了 0.1Å 噪声(类似于 ProteinMPNN),它在从头开始的骨架序列设计中表现出次优性能。 进一步研究更大的噪声添加或替代模型架构对 ESM-IF1 性能的影响可能会提供有见地的结果。 值得注意的是,我们没有在本研究中包含功能性突变预测任务,因为 ESM-IF1 在该领域已经展现出令人印象深刻的结果,并且这些任务已经在其他基准测试中得到了广泛的研究,例如 ProteinGYM (Notin 等人,2024)。 ESM3 (Hayes 等人,2024) 是最近发布的多模态蛋白质语言模型,其性能与 ESM-IF1 相当,在特定序列长度(100、300 和 400 个残基)方面具有显著优势。 我们注意到,目前 ProteinBench 中没有包含某些逆折叠方法,例如 PiFold (Gao 等人,2022) 和 CarbonDesign (Ren 等人,2024)。 我们计划在不久的将来更新它们的性能。
| Fitting Evolution Distribution | De novo backbones based sequence design | |||||||||||
| CASP | CAMEO | length 100 | length 200 | length 300 | length 400 | length 500 | ||||||
| AAR ↑ | AAR ↑ | scTM ↑ | pLDDT ↑ | scTM ↑ | pLDDT ↑ | scTM ↑ | pLDDT ↑ | scTM ↑ | pLDDT ↑ | scTM ↑ | pLDDT ↑ | |
| ProteinMPNN | 0.450 | 0.468 | 0.962 | 94.14 | 0.945 | 89.34 | 0.962 | 90.28 | 0.875 | 83.76 | 0.568 | 67.09 |
| ESM-IF1 | N/A | N/A | 0.810 | 88.83 | 0.635 | 69.67 | 0.336 | 74.36 | 0.449 | 64.59 | 0.462 | 58.97 |
| LM-Design | 0.516 | 0.570 | 0.834 | 78.45 | 0.373 | 58.41 | 0.481 | 69.86 | 0.565 | 59.87 | 0.397 | 56.35 |
| ESM3 | N/A | N/A | 0.942 | 86.60 | 0.486 | 60.69 | 0.632 | 70.78 | 0.564 | 62.63 | 0.452 | 59.37 |
3.1.2 结构设计
在本节中,我们评估蛋白质基础模型在骨架设计中的性能。 结果如表 3 所示。 我们分析的重点是生成结构在各种链长上的质量、新颖性和多样性。 基于 scTM-score 和 scRMSD 的质量指标,RFdiffusion (Watson 等人,2023b) 在 50 到 300 个氨基酸的链长范围内表现出卓越的主链设计性能。 FrameFlow (Yim 等人,2023) 在该范围内取得了第二好的成绩。 但是,我们观察到所有模型在更长的链(500 个氨基酸)上性能显着下降,scTM 分数下降超过 20%。 这种下降表明,开发用于长链主链设计的方法仍然是未来研究中的一项重要挑战。 新颖性是一个同样重要的指标,因为它衡量了一种方法探索超出已知蛋白质折叠的新结构空间的能力。 在适度质量约束下(scTM 分数 > 0.5),FoldFlow (Bose 等人,2023) 和 Genie (Lin & AlQuraishi, 2023) 在生成新结构方面表现良好。 当我们提高质量阈值(scTM 分数 > 0.8)时,Chroma (Ingraham 等人,2023) 通常在 50 到 500 个氨基酸的链长范围内表现出最佳性能。 在结构多样性方面,Chroma 在测试的链长范围内表现出色。 重要的是要注意,对于此评估,我们使用了在较小的训练集上训练的已发布的 FoldFlow 模型,该训练集包含较短的序列。 这种限制可能会导致模型架构与在整个 PDB 数据库上训练的其他方法进行不公平的比较,特别是对于更长的链长。 我们将很快更新我们的评估,以包括更多方法,例如 Foldingdiff (Wu 等人,2024a) 和 Proteous (Wang 等人,2024a)。
| length 50 | length 100 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Quality | Novelty | Diversity | Quality | Novelty | Diversity | |||||
| scTM ↑ | scRMSD ↓ | Max TM ↓ | pairwise TM ↓ | Max Clust. ↑ | scTM ↑ | scRMSD ↓ | Max TM ↓ | pairwise TM ↓ | Max Clust.↑ | |
| Native PDBs | 0.910.11 | 0.741.45 | N/A | 0.290.03 | 0.66 | 0.960.10 | 0.671.61 | N/A | 0.300.02 | 0.77 |
| RFdiffusion | 0.950.12 | 0.451.71 | 0.650.16 | 0.580.05 | 0.67 | 0.980.12 | 0.480.56 | 0.760.01 | 0.410.03 | 0.32 |
| FrameFlow | 0.910.09 | 0.580.51 | 0.750.01 | 0.680.10 | 0.39 | 0.940.08 | 0.700.70 | 0.720.01 | 0.550.08 | 0.49 |
| Chroma | 0.850.15 | 1.051.49 | 0.590.08 | 0.290.01 | 0.48 | 0.890.13 | 1.271.85 | 0.700.01 | 0.350.03 | 0.59 |
| FrameDiff(latest) | 0.850.13 | 1.001.27 | 0.670.01 | 0.350.02 | 0.64 | 0.900.08 | 1.231.02 | 0.710.08 | 0.520.05 | 0.11 |
| FoldFlow1(sfm) | 0.900.10 | 0.670.88 | 0.680.03 | 0.630.07 | 0.48 | 0.870.11 | 1.341.42 | 0.650.01 | 0.490.08 | 0.83 |
| FoldFlow1(base) | 0.790.14 | 1.191.27 | 0.660.02 | 0.530.08 | 0.76 | 0.810.15 | 1.701.95 | 0.620.01 | 0.480.07 | 0.83 |
| FoldFlow1(ot) | 0.830.16 | 1.101.53 | 0.650.02 | 0.530.08 | 0.77 | 0.830.15 | 1.601.95 | 0.640.01 | 0.480.06 | 0.81 |
| Genie | 0.570.15 | 3.122.07 | 0.570.03 | 0.320.02 | 0.90 | 0.690.17 | 3.383.04 | 0.590.01 | 0.310.02 | 0.96 |
| length 300 | length 500 | |||||||||
| Quality | Novelty | Diversity | Quality | Novelty | Diversity | |||||
| scTM ↑ | scRMSD ↓ | Max TM ↓ | pairwise TM ↓ | Max Clust. ↑ | scTM ↑ | scRMSD ↓ | Max TM ↓ | pairwise TM ↓ | Max Clust.↑ | |
| Native PDBs | 0.970.10 | 0.822.67 | N/A | 0.280.02 | 0.77 | 0.970.17 | 1.075.96 | N/A | 0.290.03 | 0.8 |
| RFdiffusion | 0.960.15 | 1.033.14 | 0.640.01 | 0.360.03 | 0.65 | 0.790.19 | 5.605.66 | 0.620.004 | 0.330.02 | 0.89 |
| FrameFlow | 0.920.15 | 1.952.76 | 0.650.01 | 0.430.07 | 0.88 | 0.610.19 | 7.924.08 | 0.610.01 | 0.400.06 | 0.92 |
| Chroma | 0.870.13 | 2.473.63 | 0.660.01 | 0.360.04 | 0.67 | 0.720.18 | 6.715.76 | 0.600.01 | 0.290.01 | 0.99 |
| FrameDiff(latest) | 0.870.12 | 2.732.69 | 0.690.00 | 0.480.04 | 0.21 | 0.630.24 | 9.5218.19 | 0.580.03 | 0.400.06 | 0.52 |
| FoldFlow1(sfm) | 0.450.11 | 9.042.52 | 0.540.01 | 0.390.04 | 1.00 | 0.370.06 | 13.041.71 | 0.530.01 | 0.370.03 | 1.00 |
| FoldFlow1(base) | 0.430.09 | 9.562.42 | 0.540.01 | 0.390.05 | 0.98 | 0.350.05 | 13.202.29 | 0.520.01 | 0.390.05 | 1.00 |
| FoldFlow1(ot) | 0.540.12 | 8.212.38 | 0.580.00 | 0.410.06 | 0.94 | 0.370.06 | 12.482.00 | 0.510.01 | 0.350.03 | 1.00 |
| Genie | 0.270.02 | 20.371.70 | 0.300.01 | 0.230.01 | 1.00 | 0.250.01 | 26.081.58 | 0.220.002 | 0.230.004 | 1.00 |
3.1.3 序列设计
| length 100 | length 200 | |||||||||
| Quality | Diversity | Novelty | Quality | Diversity | Novelty | |||||
| ppl ↓ | pLDDT ↑ | pairwise TM ↓ | Max Clust. ↑ | Max TM ↓ | ppl ↓ | pLDDT↑ | pairwise TM ↓ | Max Clust. ↑ | Max TM ↓ | |
| Native Seqs | 68.4616.50 | 0.550.19 | 0.75 | N/A | 61.9111.62 | 0.490.10 | 0.78 | N/A | ||
| Progen 2 (700M) | 8.283.87 | 64.0021.26 | 0.420.10 | 0.94 | 0.640.08 | 5.683.64 | 69.919.23 | 0.400.13 | 0.91 | 0.690.05 |
| EvoDiff | 16.891.04 | 50.2010.27 | 0.430.05 | 0.98 | 0.690.03 | 17.281.64 | 50.6616.38 | 0.360.04 | 1.00 | 0.710.02 |
| DPLM (650M) | 6.213.10 | 85.3814.20 | 0.500.20 | 0.80 | 0.740.10 | 4.612.63 | 93.543.73 | 0.540.24 | 0.70 | 0.910.004 |
| ESM3 (1.4B) | 14.792.90 | 54.2615.35 | 0.450.15 | 0.90 | 0.680.07 | 12.962.38 | 58.459.40 | 0.350.07 | 1.00 | 0.800.01 |
| length 300 | length 500 | |||||||||
| Quality | Diversity | Novelty | Quality | Diversity | Novelty | |||||
| ppl ↓ | pLDDT ↑ | pairwise TM ↓ | Max Clust. ↑ | Max TM ↓ | ppl ↓ | pLDDT↑ | pairwise TM ↓ | Max Clust. ↑ | Max TM ↓ | |
| Native Seqs | 61.4914.47 | 0.510.13 | 0.85 | N/A | 62.9512.60 | 0.510.11 | 0.78 | N/A | ||
| Progen 2 (700M) | 6.25 4.02 | 65.6920.93 | 0.420.16 | 0.93 | 0.660.06 | 4.273.60 | 61.4520.17 | 0.320.11 | 0.95 | 0.680.08 |
| EvoDiff | 17.132.00 | 45.149.95 | 0.310.03 | 1.00 | 0.680.02 | 16.513.82 | 43.145.16 | 0.310.03 | 1.00 | 0.690.02 |
| DPLM (650M) | 3.471.44 | 93.075.77 | 0.570.25 | 0.63 | 0.910.01 | 3.331.8 | 87.7311.61 | 0.430.18 | 0.85 | 0.850.04 |
| ESM3 (1.4B) | 14.592.97 | 48.0813.34 | 0.320.03 | 1.00 | 0.750.02 | 11.102.26 | 52.1710.52 | 0.300.05 | 1.00 | 0.540.03 |
在本节中,我们根据不同链长的生成序列的质量、多样性和新颖性,评估了各种蛋白质序列生成模型的性能。 评估指标包括 AlphaFold2 (AF2) 预测的 pLDDT 分数(结构合理性)、最大 TM 分数和最大聚类值(结构多样性)以及对 PDB 结构的最大 TM 分数(结构新颖性)。 我们选择了具有不同建模基础的代表性方法进行评估。 在评估的方法中,ProGen2 (Nijkamp 等人,2023) 是一个自回归蛋白质语言模型 (AR-LM),而 EvoDiff (Alamdari 等人,2023) 被设计为一个与顺序无关的自回归扩散模型 (OADM)。 DPLM (Wang 等人,2024b) 和 ESM3 (Hayes 等人,2024) 作为吸收离散扩散模型或生成掩码语言模型,共享概率基础。 值得注意的是,ESM3 是一个多模态模型,通过标记化共同学习蛋白质序列、结构和功能,超越了其他仅限于序列的方法。 对于每个模型和序列长度,我们抽取 50 个序列来评估它们的性能。
如表 4 所示,DPLM 始终显示出最高的质量分数,表明其在序列生成方面的精度更高。 然而,它的多样性指标相对较低,表明其生成的序列变化较小。 EvoDiff 虽然 pLDDT 分数较低,但在多样性方面表现出色,特别是在产生高度多样化的序列簇方面。 令人惊讶的是,ESM3,一种多模态蛋白质 LM,在序列生成方面显示出较低的 pLDDT,同时保持着竞争力的多样性,尤其是在生成新序列方面。 ProGen2 在质量和多样性之间取得平衡,提供中等 pLDDT 分数以及令人满意的多样性和新颖性。 此模型可有效生成既多样化又接近已知结构的序列,具体取决于特定应用需求。 对于不同的链长,所有模型在其性能指标方面通常都表现出一致的趋势。 随着链长的增加,一些模型生成的序列质量略有下降,特别是对于 EvoDiff 和 ESM3。 这表明在链长增加时保持高序列质量是一个挑战。 其中,DPLM 在所有长度上都表现出稳健的性能,即使对于更长的序列也能保持高 pLDDT。 总体而言,DPLM 擅长生成高度结构化的蛋白质序列,而 EvoDiff 和 ESM3 更适合更高的多样性和新颖性,ProGen2 在各个指标上都提供均衡的性能。
3.1.4 结构和序列协同设计
∗: 我们已尽力根据其各自代码库中的说明,使用公开可用的模型权重来复制所有模型。 但是,某些结果可能与原始研究中报告的结果不同。 我们欢迎任何反馈和更正,以帮助我们在未来及时更新。
| length 100 | length 200 | |||||||
| Quality | Diversity | Novelty | Quality | Diversity | Novelty | |||
| scTM ↑ | scRMSD ↓ | Max Clust. ↑ | Max TM ↓ | scTM ↑ | scRMSD ↓ | Max Clust. ↑ | Max TM ↓ | |
| Native PDBs | 0.910.11 | 2.983.49 | 0.75 | N/A | 0.880.09 | 3.243.77 | 0.77 | N/A |
| ProteinGenerator | 0.910.08 | 3.753.39 | 0.24 | 0.73 | 0.880.09 | 6.244.10 | 0.25 | 0.72 |
| ProtPardelle* | 0.560.12 | 12.91.88 | 0.57 | 0.66 | 0.640.11 | 13.672.80 | 0.10 | 0.69 |
| Multiflow | 0.960.04 | 1.100.71 | 0.33 | 0.71 | 0.950.04 | 1.611.73 | 0.42 | 0.71 |
| ESM3* | 0.720.19 | 13.8010.51 | 0.64 | 0.41 | 0.630.20 | 21.1816.19 | 0.63 | 0.61 |
| length 300 | length 500 | |||||||
| Quality | Diversity | Novelty | Quality | Diversity | Novelty | |||
| scTM ↑ | scRMSD ↓ | Max Clust. ↑ | Max TM ↓ | scTM ↑ | scRMSD ↓ | Max Clust. ↑ | Max TM ↓ | |
| Native PDBs | 0.920.12 | 3.944.95 | 0.75 | N/A | 0.900.14 | 9.647.05 | 0.80 | N/A |
| ProteinGenerator | 0.810.14 | 9.264.13 | 0.22 | 0.71 | 0.690.17 | 17.005.52 | 0.18 | 0.73 |
| ProtPardelle* | 0.690.08 | 14.913.45 | 0.04 | 0.72 | 0.440.12 | 43.159.86 | 0.60 | 0.69 |
| Multiflow | 0.960.06 | 2.143.24 | 0.58 | 0.71 | 0.950.07 | 2.713.65 | 0.62 | 0.71 |
| ESM3* | 0.590.21 | 25.520.68 | 0.52 | 0.73 | 0.640.20 | 26.7221.08 | 0.46 | 0.78 |
在本节中,我们考察了蛋白质结构序列协同生成的性能,这是一个最近在研究界引起广泛关注的主题。 我们检查了 ProteinGenerator (Lisanza 等人,2023)、ProtPardelle (Chu 等人,2024)、Multiflow (Campbell 等人,) 和 ESM3 (Hayes 等人,2024) 在不同长度下的性能。 性能评估使用与主干生成中使用的指标类似的指标。 然而,请注意,这里的质量是关于结构序列兼容性,使用 scTM 和 scRMSD 测量设计的序列折叠成相应的设计的结构的程度。 主要区别在于,协同设计模型的任务是同时生成序列和结构,而主干设计模型需要额外的逆折叠模型(例如 ProteinMPNN)来设计序列。 用于评估的其他指标包括多样性(最大簇)和新颖性(PDB 的最大 TM 分数)。
如表 5 所示,ProteinGenerator 和 Multiflow 在所有序列长度上始终展现出强大的结构序列兼容性性能,具有较高的 scTM 分数(高达 0.96±0.06)和相对较低的 scRMSD 值,表明生成的序列具有优异的结构质量。 ProteinGenerator 尤其擅长较短的长度,在质量和多样性指标之间表现出平衡的性能。 Multiflow 即使在序列长度增加的情况下也能保持高性能,表明它具有鲁棒性,始终保持较高的 scTM 分数和较低的 scRMSD 值,这表明它能够生成高质量的结构。 另一方面,ProtPardelle 和 ESM3 在序列长度增加时性能下降,如其较低的 scTM 分数和非常高的 scRMSD 值所示,表明它难以保持较长序列的结构质量。 总之,这些发现表明,虽然 ProteinGenerator 和 Multiflow 是用于生成不同长度的高质量蛋白质结构的有效模型,但 Multiflow 在所有测试长度上都特别鲁棒。
3.1.5 基序-支架
在本节中,我们评估了各种基序-支架方法在 Watson 等人(2023b) 和 Yim 等人(2024) 中使用的不同支架上的性能,重点关注它们在设计支架结构方面的有效性。 本次评估的主要目标是比较基于结构和基于序列的方法在生成可设计支架方面的效力。 对于纯粹的基于序列的方法,例如 EvoDiff (Alamdari 等人,2023) 和 DPLM (Wang 等人,2024b),我们使用 ESMFold 预测其设计的基序-支架序列的结构。
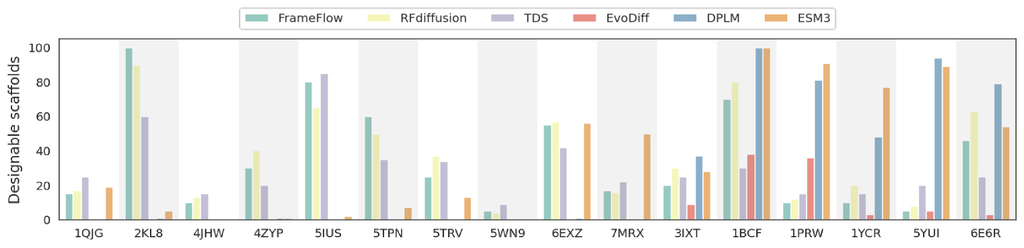
图 3 显示了测试方法之间广泛的性能水平,每种方法都表现出独特的优势和劣势,具体取决于特定的支架环境。 值得注意的是,基于结构的方法,如 RFdiffusion (Watson 等人,2023b)、TDS (Wu 等人,2024b) 和 FrameFlow (Yim 等人,2024) 在大多数情况下始终表现良好,其中 RFdiffusion 在生成大量可设计支架方面表现出特别的稳健性。 这表明基于结构的方法在捕捉成功支架设计所需的复杂结构细节方面非常有效。 相反,像 EvoDiff 和 DPLM 这样的基于序列的方法表现出不同的性能,在主要受进化约束控制的某些支架中表现出色,但在具有更复杂结构基序的其他支架中表现不佳。 这种可变性可能反映了它们在识别和适应特定结构特征方面的局限性。
有趣的是,ESM3 (Hayes 等人,2024) 是最新的基于序列的方法和多模态语言模型,能够通过结构标记感知三级特征,在大多数情况下生成可设计支架方面表现出具有竞争力的性能。 它的性能与更先进的基于结构的模型相当。 这表明像 ESM3 这样的多模态语言模型可以有效地将结构功能集成到统一的语言建模框架中,使其成为支架设计的通用工具。 然而,ESM3 并不在所有情况下始终如一地接近基于结构的方法,这表明虽然多模态蛋白质语言模型很有前景,但还需要进一步改进和优化才能在不同的结构挑战中实现更一致的性能。
总之,我们的发现强调,目前还没有一个单一模型在所有支架中都能普遍出色,这突出了选择与特定设计目标相符的基序-支架方法的重要性。 未来研究应探索这些方法的整合,以利用它们各自的优势,这可能导致更强大和更通用的支架设计能力。
3.1.6 抗体设计
| Accuracy | Functionality | Specificity | |||||
| AAR ↑ | RMSD ↓ | TM-score ↑ | Binding Energy ↓ | SeqSim-outer ↓ | SeqSim-inner ↑ | PHR ↓ | |
| RAbD (natural) | 100.00% | 0.00 | 1.00 | -15.33 | 0.26 | N/A | 45.78% |
| HERN | 33.17% | 9.86 | 0.16 | 1242.77 | 0.41 | N/A | 39.83% |
| MEAN | 33.47% | 1.82 | 0.25 | 263.90 | 0.65 | N/A | 40.74% |
| dyMEAN | 40.95% | 2.36* | 0.36 | 889.28 | 0.58 | N/A | 42.04% |
| dyMEAN-FixFR | 40.05%1.06 | 2.370.03 | 0.350.01 | 612.7556.03 | 0.60 | 0.96 | 43.75%2.24 |
| DiffAb | 35.04%8.36 | 2.530.60 | 0.370.06 | 489.42499.76 | 0.37 | 0.45 | 40.68%10.65 |
| AbDPO | 31.29%7.29 | 2.793.01 | 0.350.06 | 116.06186.06 | 0.38 | 0.60 | 69.69%8.49 |
| AbDPO++ | 36.25%7.95 | 2.480.59 | 0.350.06 | 223.73281.7 | 0.39 | 0.54 | 44.51%9.55 |
| Rationality | |||||||
| CN-score ↑ | Clashes-inner ↓ | Clashes-outer ↓ | SeqNat↑ | Total Energy ↓ | scRMSD ↓ | ||
| RAbD (natural) | 50.19 | 0.07 | 0.00 | -1.74 | -16.76 | 1.77 | |
| HERN | 0.04 | 0.04 | 3.25 | -1.47 | 5408.74 | 9.89 | |
| MEAN | 1.33 | 11.65 | 0.29 | -1.83 | 1077.32 | 2.77 | |
| dyMEAN | 1.49 | 9.15 | 0.47 | -1.79 | 1642.65 | 2.11 | |
| dyMEAN-FixFR | 1.141.71 | 8.880.55 | 0.480.12 | -1.820.10 | 1239.29113.84 | 2.480.24 | |
| DiffAb | 2.022.83 | 1.841.35 | 0.190.31 | -1.880.47 | 495.69350.96 | 2.570.77 | |
| AbDPO | 1.332.31 | 4.141.84 | 0.100.24 | -1.990.34 | 270.12217.45 | 2.793.25 | |
| AbDPO++ | 2.343.20 | 1.661.28 | 0.080.20 | -1.780.43 | 338.14266.48 | 2.500.75 | |
在本节中,我们选择了五种抗原特异性抗体设计方法 (HERN (Jin 等人,2022),MEAN (Kong 等人,2022),dyMEAN (Kong 等人,2023),DiffAb (Luo 等人,2022),AbDPO (Zhou 等人,2024)) 以及它们的两种变体 (根据附录 B.1.2 实现的 dyMEAN-FixFR 和 AbDPO++),总共七种方法,以评估它们在针对给定抗原的 CDR-H3 生成中的性能。 所有方法都使用相应论文中报告的参数在相同的数据集上进行训练,并在 RAbD 数据集 (Adolf-Bryfogle 等人,2018) 中的 55 个测试案例的公共集合上进行测试,详情请参见附录 B.1.3。 值得注意的是,dyMEAN-FixFR 不是 dyMEAN 的官方变体;我们修改了 dyMEAN,使其任务设置与其他方法一致,并允许它为同一个抗原生成不同的抗体。 最终的评估结果如表格 6 所示。 对于每个评估指标,我们突出显示了 最佳 性能 (粗体) 和 次佳 性能 (下划线),每个指标的详细实现可以在附录 B.1.4 中找到。
在 准确性 评估中,dyMEAN 和 MEAN 在序列和结构方面取得了最佳性能 (最高 AAR 和最低 RMSD),而 DiffAb 在 TM-score 方面表现最佳。 然而,考虑到多个评估指标,这些方法的总体性能并不出色。 此外,除了 HERN 之外,其他方法之间没有显著的性能差异。
在 功能性 评估中,所有方法生成的抗体与给定抗原的结合能都显著高于天然抗体。 AbDPO 和 AbDPO++ 在结合能方面取得了所有方法中最好的性能。
在抗体的 特异性 评估中,我们主要观察了针对不同抗原的抗体之间的序列相似性 (SeqSim-outer) 和生成的抗体中疏水残基的比例 (PHR)。 前者指标表明该方法是否可以设计出针对给定抗原的特异性抗体,而后者反映了由于疏水性高而可能发生的非特异性结合。
-
•
在 SeqSim-outer 中,我们注意到 MEAN 和 dyMEAN 为不同的抗原生成了高度相似的序列(我们测试集中最大的 SeqSim-outer 为 0.79,表明所有抗体差异仅来自长度变化)。 这表明它们出色的 AAR 可能源于学习抗体序列中的高频模式,根据这些模式为不同的抗原生成抗体。 相比之下,DiffAb 和 AbDPO 表现最佳。
-
•
对于能够为相同抗原生成不同抗体的方法,我们还测量了为相同抗原生成的抗体之间的序列相似性 (SeqSim-inner)。 我们预计为相同抗原生成的抗体将更加相似。 在这方面,dyMEAN-FixFR 和 AbDPO 表现最佳。 然而,dyMEAN-FixFR 的 0.96 SeqSim-inner 表明,尽管在模型初始化过程中引入了随机性,但最终的序列生成几乎没有差异。 此外,DiffAb 在 SeqSim-outer 中表现最佳,但为相同抗原生成的抗体相似度较低,这表明序列生成可能存在欠拟合。 考虑到两种类型的 SeqSim,AbDPO 取得了最佳性能。
-
•
在 PHR 中,HERN 和 dyMEAN 表现最佳,但总体而言,几乎所有方法的性能都优于天然抗体。 只有 AbDPO 生成了过多的疏水残基,降低了特异性。 然而,它的变体 AbDPO++ 很好地控制了 PHR,在所有方法中与天然抗体非常匹配。
Rationality 评估包括三个方面:结构合理性、序列合理性和结构与序列联合合理性。
-
•
在结构合理性方面,我们关注肽键长度符合天然肽键长度分布的评分 (CN-score),生成结构中潜在内部碰撞的数量 (Clashes-inner) 以及生成结构与其他部分之间的碰撞 (Clashes-outer)。 很明显,生成的抗体中普遍存在非理性结构,但总的来说,基于扩散的方法表现更好。 AbDPO++ 和 DiffAb 在所有方法中取得了最佳性能。 HERN 和 MEAN/dyMEAN 在 碰撞(内部/外部)方面表现出不同的趋势,这与我们对生成的样本的观察结果相符。 HERN 倾向于生成较大的 CDR-H3 结构,导致内部碰撞较少,但与抗原的碰撞更多,而 MEAN/dyMEAN 倾向于生成较小的 CDR-H3 结构。
-
•
在序列合理性方面,我们使用 AntiBERTy 的逆困惑度 (Ruffolo 等人,2021) 来表示序列的自然性,即 SeqNat,结果表明 HERN 的表现最好,这可能是因为 HERN 是唯一一个自回归模型。 AbDPO++ 取得了第二好的成绩,并且最接近天然抗体。
-
•
在结构和序列的联合评估中,我们主要从两个方面关注生成的结构和序列之间的一致性:物理能量和结构预测。 在物理能量方面,我们计算了生成的 CDR-H3 的总能量 (总能量),这将受到侧链引起的碰撞的严重影响,从而反映出生成的结构和序列之间的非理性。 在这种与能量相关的指标中,AbDPO 和 AbDPO++ 在所有方法中表现最好。 从结构预测的角度来看,我们使用 IgFold (Ruffolo 等人,2023) 来预测生成的序列的结构,在以抗原为条件的情况下进行后优化,并计算预测结构和生成的结构之间的 CA-RMSD (scRMSD)。 dyMEAN 和 dyMEAN-FixFR 在 scRMSD方面表现最好。 尽管这两个指标都反映了序列和结构之间的一致性,但它们关注的是不同的方面。 此外,能量计算和结构预测都存在固有误差,因此不同方法的性能在这两个指标上可能并不一致。
总之,评估抗体设计方法包括各个方面,仅使用几个指标将严重误导研究人员对模型性能的理解。 此外,我们必须认识到,没有一种方法在所有方面都优于其他方法,并且所有方法与天然抗体相比都存在显着差距。 这些差异可能是由于结构化数据的严重缺乏造成的,导致模型专注于某些序列模式或结构。 此外,大多数模型不执行抗体和抗原的原子级建模,从而无法进行准确的相互作用建模。 必须开发新的任务范式来克服抗体设计中当前的挑战。 尽管如此,AbDPO++ 通过利用合成数据并与各种属性对齐,在几乎所有方面都取得了所有方法中最好的性能之一,没有表现出明显的弱点。
3.2 蛋白质构象预测
在 ProteinBench 的第二部分,我们重点关注构象预测,这是另一类跨模态任务,旨在根据蛋白质序列预测蛋白质结构(构象)。 虽然当前的模型基于与设计任务不同的研究工作,但预测蛋白质构象的能力可以洞察模型对蛋白质结构的物理和动力学的理解。 这种能力对于未来的蛋白质基础模型完全理解、预测和设计体现关键序列-结构-功能关系的蛋白质至关重要。
构象预测模型的开发仍处于起步阶段,目前只提出了少数探索性方法。 这些方法之间的全面比较尚未进行。 据我们所知,这是对当前构象预测模型的首次基准研究,其中包括迄今为止提出的主要策略:(1)扰乱折叠模型的序列输入 (Del Alamo et al., 2022; Wayment-Steele et al., 2024);(2)通过仅结构扩散模型扰乱蛋白质结构 (Lu et al., 2024);(3)在来自实验或模拟的大规模结构数据上训练生成模型 (Jing et al., 2023; 2024; Wang et al., 2024c; Zheng et al., 2024);(4)使用物理模型改进构象采样 (Zheng et al., 2024; Wang et al., 2024c)。
3.2.1 蛋白质折叠:单状态预测
虽然大多数折叠模型,如 AlphaFold2 (Jumper et al., 2021) 和 ESMFold (Lin et al., 2023) 本质上不是生成性的,但我们仍然将它们视为构象预测的“蛋白质基础模型”,因为(1)它们在大量的结构和/或序列数据上进行训练;(2)它们在理解序列-结构关系方面发挥了基础性作用;(3)它们与 ESM2 (Rives et al., 2021) 和 AlphaFold3 (Abramson et al., 2024) 等基础模型密切相关,并作为构象预测模型的关键构建块 (Jing et al., 2024; Wang et al., 2024c)。 因此,在讨论蛋白质构象预测时,对其性能进行基准测试至关重要。
在表 7 中,我们总结了折叠模型在 CAMEO2022 上的结果。 对于 AlphaFold2、OpenFold 和 EigenFold,我们预测了五个结构,并根据模型的内部置信度得分报告了最佳结构。 与之前的报道一致 (Jing et al., 2023),基于多序列比对(MSA)的折叠模型(AlphaFold2、OpenFold、RosettaFold2)优于基于蛋白质语言模型的折叠模型(ESMFold、EigenFold)。 AlphaFold2 及其忠实复制品 OpenFold 在所有准确性指标上都表现出最佳性能。 预测结构的质量在模型之间是可比的(EigenFold 除外),具有最小的残基间碰撞或键断裂。 EigenFold (Jing 等人,2023) 是第一个声称同时具备蛋白质折叠和构象预测能力的扩散生成模型之一;然而,其性能可能受到几个设计因素的限制:它基于 OmegaFold Wu 等人 (2022),使用仅包含α碳的粗粒度表示,并且模型尺寸较小,只有 572K 可训练参数。 总之,虽然 MSA 搜索非常耗时,但 AlphaFold2 和 OpenFold 在蛋白质折叠的准确性和结构质量方面仍取得了最佳性能,为蛋白质构象预测提供了坚实的基础。
| Accuracy | Quality | ||||||
|---|---|---|---|---|---|---|---|
| TM-score ↑ | RMSD ↓ | GDT-TS ↑ | lDDT ↑ | CA clash (%) ↓ | CA break (%) ↓ | PepBond break (%) ↓ | |
| AlphaFold2 | 0.871/0.952 | 3.21/1.64 | 0.860/0.921 | 0.904/0.933 | 0.3/0.0 | 0.0/0.0 | 4.8/4.1 |
| OpenFold | 0.870/0.947 | 3.21/1.59 | 0.856/0.913 | 0.899/0.933 | 0.4/0.0 | 0.0/0.0 | 2.0/1.7 |
| RoseTTAFold2 | 0.859/0.941 | 3.52/1.75 | 0.845/0.903 | 0.892/0.926 | 0.3/0.0 | 0.2/0.0 | 5.5/4.0 |
| ESMFold | 0.847/0.929 | 3.98/2.10 | 0.826/0.881 | 0.870/0.907 | 0.3/0.0 | 0.0/0.0 | 4.7/3.4 |
| EigenFold* | 0.743/0.823 | 7.65/3.73 | 0.703/0.781 | 0.737/0.810 | 8.0/4.6 | 0.5/0.0 | N/A |
3.2.2 多状态预测
本节评估了五种构象预测模型及其变体的预测多个构象状态的性能:EigenFold Jing 等人 (2023)、MSA-subsampling (Del Alamo 等人,2022)、Str2Str (Lu 等人,2024)、AlphaFlow/ESMFlow Jing 等人 (2024) 和 ConfDiff (Wang 等人,2024c)。 这里重点介绍了这些方法的关键区别。 MSA-subsampling 通过减少输入 MSA 的数量(称为“深度”)来扰乱 OpenFold 的模型输入,从而允许对蛋白质的不同构象进行采样。 Str2Str 使用了一个仅限结构的扩散模型(即骨架设计模型)来生成构象,方法是通过前向-后向扩散过程扰乱初始折叠预测。 扰动程度由最大扩散时间 控制,并通过在不同扩散时间 采样结构来生成集合输出。 EigenFold、AlphaFlow/ESMFlow 和 ConfDiff 通过使用 PDB 中的结构数据微调折叠模型来训练扩散或流模型,采用类似的方法。 虽然 AlphaFlow/ESMFlow 对原始模型的所有层进行微调,但 EigenFold 和 ConfDiff 仅使用来自折叠模型的预训练表示,并训练一个轻量级模块来进行评分或结构预测。 此外,AlphaFlow/ESMFlow 和 ConfDiff 都提供了进一步在最新的 MD构象数据集 (Vander Meersche et al., 2024) 上微调的版本,用“-MD”后缀表示。 ConfDiff (Wang et al., 2024c) 引入了两种指导技术来改进构象采样:(1) 无分类器指导,它将序列条件的构象模型与无条件的(仅结构)模型结合起来探索构象空间(ConfDiff-ClsFree);以及 (2) 能量/力指导,它通过辅助预测模块对中间能量/力进行指导,将采样引导到具有较低势能的区域(ConfDiff-Energy/Force)。 但是,这种物理预测模块是特定于数据集的,并且仅适用于 BPTI 数据集。
| RMSDens ↓ | RMSD Cluster 3 ↓ | Diversity | Quality | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N=10 | N=100 | N=500 | N=1000 | N=10 | N=100 | N=500 | N=1000 |
|
|
|
|
|||||||||
| EigenFold | 1.560.02 | 1.500.01 | 1.470.01 | 1.460.00 | 2.540.03 | 2.480.01 | 2.460.01 | 2.460.01 | 0.85 | 1.4 | 4.3 | N/A | ||||||||
| MSA-depth256 | 1.570.01 | 1.540.01 | 1.520.00 | 1.520.01 | 2.510.02 | 2.470.02 | 2.450.02 | 2.450.02 | 0.20 | 0.0 | 0.0 | 9.2 | ||||||||
| MSA-depth64 | 1.600.02 | 1.540.01 | 1.510.01 | 1.500.01 | 2.480.03 | 2.400.04 | 2.350.04 | 2.330.03 | 0.55 | 0.0 | 0.0 | 7.9 | ||||||||
| MSA-depth32 | 1.670.05 | 1.530.04 | 1.450.04 | 1.410.02 | 2.390.15 | 2.210.15 | 1.930.15 | 1.870.06 | 2.14 | 0.6 | 0.0 | 10.6 | ||||||||
| Str2Str-ODE () | 2.360.10 | 2.190.06 | 2.100.02 | 2.080.01 | 3.030.17 | 2.680.12 | 2.600.05 | 2.560.02 | 1.86 | 0.0 | 0.0 | 13.9 | ||||||||
| Str2Str-SDE () | 2.830.23 | 2.480.11 | 2.280.04 | 2.250.03 | 3.420.32 | 2.920.28 | 2.520.14 | 2.480.13 | 3.60 | 0.3 | 0.0 | 16.0 | ||||||||
| AlphaFlow-PDB | 1.530.02 | 1.450.01 | 1.420.01 | 1.410.01 | 2.480.04 | 2.430.02 | 2.410.02 | 2.400.01 | 0.86 | 0.0 | 0.0 | 13.2 | ||||||||
| AlphaFlow-MD | 1.740.09 | 1.510.04 | 1.450.02 | 1.430.02 | 2.440.06 | 2.320.06 | 2.280.04 | 2.240.00 | 1.26 | 0.0 | 0.1 | 26.2 | ||||||||
| ESMFlow-PDB | 1.610.04 | 1.490.02 | 1.440.01 | 1.420.01 | 2.470.05 | 2.410.03 | 2.370.03 | 2.350.01 | 0.74 | 0.0 | 0.0 | 6.0 | ||||||||
| ESMFlow-MD | 1.660.07 | 1.500.04 | 1.410.03 | 1.400.02 | 2.490.06 | 2.290.09 | 2.200.04 | 2.180.03 | 1.17 | 0.0 | 0.0 | 14.3 | ||||||||
| ConfDiff-Open-ClsFree | 1.650.05 | 1.480.05 | 1.410.04 | 1.370.03 | 2.560.05 | 2.300.23 | 2.160.20 | 2.030.13 | 1.77 | 0.5 | 0.0 | 5.5 | ||||||||
| ConfDiff-Open-MD | 1.640.04 | 1.500.02 | 1.440.02 | 1.420.02 | 2.490.08 | 2.390.05 | 2.320.03 | 2.310.02 | 1.37 | 0.2 | 0.0 | 4.6 | ||||||||
| ConfDiff-ESM-ClsFree | 1.580.05 | 1.450.02 | 1.410.01 | 1.390.00 | 2.500.05 | 2.390.03 | 2.350.03 | 2.330.02 | 1.52 | 0.5 | 0.0 | 7.5 | ||||||||
| ConfDiff-ESM-MD | 1.610.03 | 1.470.02 | 1.420.01 | 1.400.01 | 2.450.08 | 2.320.06 | 2.260.04 | 2.240.01 | 1.42 | 0.1 | 0.0 | 5.0 | ||||||||
| ConfDiff-ESM-Energy | 1.630.06 | 1.470.01 | 1.430.01 | 1.420.01 | 2.550.07 | 2.430.04 | 2.410.02 | 2.400.01 | 1.26 | 0.1 | 0.0 | 7.5 | ||||||||
| ConfDiff-ESM-Force | 1.580.06 | 1.440.03 | 1.370.02 | 1.360.01 | 2.450.09 | 2.330.07 | 2.230.06 | 2.220.06 | 1.76 | 0.1 | 0.0 | 8.9 | ||||||||
在表 8 中,我们总结了预测 BPTI 的五个结构簇的结果。 具体而言,五个簇的整体准确性(RMSDens)和最难采样的 Cluster 3 的准确性(RMSD Cluster 3)通过在不同的样本大小下引导来评估。 具有无分类器指导的 ConfDiff 模型在大多数样本大小上都展现出了最佳的整体准确性(RMSDens)表现,突出了其在 BPTI 上相比于在 MD 构象数据上微调的更高效性。 与 Wang et al. (2024c) 一致,ConfDiff-ESM-Force 达到了最高的整体准确性,表明结合物理信息可以改善高精度构象的采样。 对于采样 Cluster 3 的任务,MSA 子采样尽管是一种简单的方法,但能够生成最有可能捕获这种遥远状态的构象。 随着 MSA 深度的降低,样本多样性增加,使模型能够更接近 Cluster 3 进行采样。 相比之下,Str2Str 模型在这项任务上的表现并不出色,这可能是因为仅结构的方法无法保证扰动后的结构仍然忠实于提供的序列,导致整体性能下降。 EigenFold 还显示出较低的差异性,这可能会限制其在采样不同构象方面的有效性。 AlphaFlow/ESMFlow 也展现出具有竞争力的性能。 在 MD 数据集上微调提供了更高的多样性,并改善了与 PDB 训练的基模型相比,对 Cluster 3 进行采样的准确性。 然而,我们还观察到,由于在 MD 构象数据上微调而导致的质量下降更为明显,特别是肽键断裂的频率在残基之间增加。
| Accuracy | Diversity | Quality | ||||||
|---|---|---|---|---|---|---|---|---|
| apo-TM ↑ | holo-TM ↑ | TMens ↑ | Pairwise TM | CA clash % ↓ | CA break% ↓ |
|
||
| apo model | 1.000/1.000 | 0.790/0.821 | 0.895/0.910 | N/A | N/A | N/A | N/A | |
| EigenFold | 0.831/0.862 | 0.864/0.900 | 0.847/0.874 | 0.907/0.958 | 3.6/1.2 | 0.3/0.1 | N/A | |
| MSA-depth256 | 0.845/0.882 | 0.889/0.936 | 0.867/0.894 | 0.978/0.993 | 0.2/0.0 | 0.0/0.0 | 4.6/4.0 | |
| MSA-depth64 | 0.844/0.877 | 0.883/0.927 | 0.863/0.906 | 0.950/0.980 | 0.2/0.0 | 0.0/0.0 | 5.7/5.0 | |
| MSA-depth32 | 0.824/0.865 | 0.857/0.905 | 0.841/0.882 | 0.864/0.924 | 0.2/0.0 | 0.0/0.0 | 8.9/7.3 | |
| Str2Str-ODE () | 0.762/0.791 | 0.778/0.816 | 0.770/0.794 | 0.954/0.956 | 0.2/0.0 | 0.0/0.0 | 14.0/12.4 | |
| Str2Str-ODE () | 0.766/0.797 | 0.781/0.818 | 0.774/0.797 | 0.872/0.871 | 0.2/0.0 | 0.0/0.0 | 14.7/12.9 | |
| Str2Str-SDE () | 0.682/0.703 | 0.693/0.717 | 0.688/0.712 | 0.760/0.748 | 0.2/0.1 | 1.5/1.5 | 22.6/21.3 | |
| Str2Str-SDE () | 0.680/0.685 | 0.689/0.718 | 0.684/0.697 | 0.639/0.604 | 0.2/0.1 | 1.4/1.4 | 21.1/19.6 | |
| AlphaFlow-PDB | 0.855/0.896 | 0.891/0.942 | 0.873/0.900 | 0.924/0.955 | 0.3/0.0 | 0.0/0.0 | 6.6/6.7 | |
| AlphaFlow-MD | 0.857/0.888 | 0.863/0.913 | 0.860/0.892 | 0.894/0.918 | 0.2/0.0 | 0.0/0.0 | 20.8/20.6 | |
| ESMFlow-PDB | 0.849/0.878 | 0.882/0.924 | 0.866/0.900 | 0.935/0.952 | 0.3/0.1 | 0.0/0.0 | 4.8/4.7 | |
| ESMFlow-MD | 0.851/0.882 | 0.864/0.908 | 0.858/0.890 | 0.897/0.922 | 0.1/0.0 | 0.0/0.0 | 10.9/10.9 | |
| ConfDiff-Open-ClsFree | 0.838/0.886 | 0.879/0.927 | 0.859/0.885 | 0.870/0.898 | 0.8/0.6 | 0.0/0.0 | 5.8/5.6 | |
| ConfDiff-Open-MD | 0.839/0.881 | 0.874/0.918 | 0.857/0.890 | 0.863/0.892 | 0.4/0.2 | 0.0/0.0 | 6.8/6.8 | |
| ConfDiff-ESM-ClsFree | 0.837/0.883 | 0.864/0.907 | 0.850/0.887 | 0.846/0.869 | 0.7/0.6 | 0.0/0.0 | 4.6/4.5 | |
| ConfDiff-ESM-MD | 0.836/0.877 | 0.862/0.908 | 0.849/0.892 | 0.846/0.875 | 0.3/0.2 | 0.0/0.0 | 4.1/4.0 | |
接下来,我们转向一个更大的数据集 apo,其中包括 91 种具有配体结合诱导构象变化的蛋白质(表格 9)。 在这项任务中,模型需要预测未结合 (apo) 和结合 (holo) 结构。 有趣的是,我们发现性能最佳的模型是最接近折叠模型的模型(例如,MSA-depth256、AlphaFlow-PDB)。 尽管使用了一个小的扰动级别 (),但 Str2Str 并没有准确预测 apo 或 holo 结构。 提高样本多样性的策略,例如降低 MSA 深度、在 MD 构象数据上微调或使用无分类器引导,通常不会改善(有时甚至会损害)TMens 分数。 此外,我们还包含了一个始终预测完美 apo 结构的基线模型(apo 模型),该模型的 TMens 分数高于当前模型。 这些发现表明,强大的折叠模型可以提高采样质量,但当前的性能难以超越“完美”的折叠模型,因为现有蛋白质构象模型仍然难以解决 apo-holo 构象挑战。
在多态预测任务中,我们观察到 MSA 子采样、无分类器引导、力引导以及在 MD 构象数据上训练等策略提高了 BPTI 数据集上样本的多样性和准确性。 然而,大多数这些策略未能改善 apo-holo 构象变化的双态预测任务
3.2.3 分布预测
在这项最终任务中,我们对 ATLAS 测试集上的模型进行基准测试,该测试集包含 82 种蛋白质,并重点关注每个模型恢复经典蛋白质分子动力学模拟中观察到的构象分布的能力。 结果总结在表 10 中。 为了比较,我们还包括了以下参考性能:(1) 来自 MD 生成的结构的 i.i.d. 样本 (MD iid);(2) 250 个连续样本,相当于 2.5 纳秒的模拟时间 (MD 2.5 ns)。
总体而言,经过训练从序列中采样蛋白质构象的生成模型 (AlphaFlow/ESMFlow、ConfDiff) 在灵活性预测、分布精度和系综可观察量的几乎所有准确度指标上,都比基于扰动的模型 (MSA 子采样和 Str2Str) 表现得更好。 在我们的实验中,调整 Str2Str 的扰动水平(最大前向时间 )和 MSA 子采样(MSA 深度)并没有改善与分布相关的指标,这表明仅靠扰动可能不足以准确捕捉样本分布。 我们确定了两个因素始终可以提高 AlphaFlow/ESMFlow 和 ConfDiff 的模型性能:(1) 选择一个强大的基础折叠模型(例如,AlphaFold 或 OpenFold),虽然这可能会降低样本多样性,但可以改善与分布相关的指标;(2) 在 MD 构象数据上进行微调,这进一步增强了模型预测目标分布的能力。 后者再次强调了通过监督方法将模型的分布与目标分布对齐的重要性,而不是仅仅依靠构象探索策略(例如,无分类器引导),来准确地预测分布。 此外,结果与之前关于多样性、预测性能和样本质量之间权衡的任务一致:例如,虽然在 MD 构象数据上进行微调可以提高 AlphaFlow/ESMFlow 的样本多样性和预测性能,但它也会显着提高这些模型中肽键断裂的水平。
虽然当前的构象预测模型在捕捉动力学相关特征和近似构象分布方面已经显示出可喜的迹象,但重要的是要注意,这些模型的性能与经典 MD 模拟的性能之间仍然存在明显的差距,即使是在短模拟时间(例如,2.5 ns)下也是如此。 尚未实现与从 MD 构象样本中采样 i.i.d. 相当的性能。
| Diversity | Flexibility: Pearson on | Distributional accuracy | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
*RMSF |
|
|
|
|
|
|
|
||||||||||||||||
| MD iid | 2.76 | 1.63 | 0.96 | 0.97 | 0.99 | 0.71 | 0.76 | 0.70 | 93.9 | |||||||||||||||
| MD 2.5 ns | 1.54 | 0.98 | 0.89 | 0.85 | 0.85 | 2.21 | 1.57 | 1.93 | 36.6 | |||||||||||||||
| EigenFold | 5.96 | N/A | -0.04 | N/A | N/A | N/A | 2.35 | 7.96 | 12.2 | |||||||||||||||
| MSA-depth256 | 0.84 | 0.53 | 0.25 | 0.34 | 0.59 | 3.63 | 1.83 | 2.90 | 29.3 | |||||||||||||||
| MSA-depth64 | 2.03 | 1.51 | 0.24 | 0.30 | 0.57 | 4.00 | 1.87 | 3.32 | 18.3 | |||||||||||||||
| MSA-depth32 | 5.71 | 7.96 | 0.07 | 0.17 | 0.53 | 6.12 | 2.50 | 5.67 | 17.1 | |||||||||||||||
| Str2Str-ODE (t=0.1) | 1.66 | N/A | 0.13 | N/A | N/A | N/A | 2.12 | 4.42 | 6.1 | |||||||||||||||
| Str2Str-ODE (t=0.3) | 3.15 | N/A | 0.12 | N/A | N/A | N/A | 2.23 | 4.75 | 9.8 | |||||||||||||||
| Str2Str-SDE (t=0.1) | 4.74 | N/A | 0.10 | N/A | N/A | N/A | 2.54 | 8.84 | 9.8 | |||||||||||||||
| Str2Str-SDE (t=0.3) | 7.54 | N/A | 0.00 | N/A | N/A | N/A | 3.29 | 12.28 | 7.3 | |||||||||||||||
| AlphaFlow-PDB | 2.58 | 1.20 | 0.27 | 0.46 | 0.81 | 2.96 | 1.66 | 2.60 | 37.8 | |||||||||||||||
| AlphaFlow-MD | 2.88 | 1.63 | 0.53 | 0.66 | 0.85 | 2.68 | 1.53 | 2.28 | 39.0 | |||||||||||||||
| ESMFlow-PDB | 3.00 | 1.68 | 0.14 | 0.27 | 0.71 | 4.20 | 1.77 | 3.54 | 28.0 | |||||||||||||||
| ESMFlow-MD | 3.34 | 2.13 | 0.19 | 0.30 | 0.76 | 3.63 | 1.54 | 3.15 | 25.6 | |||||||||||||||
| ConfDiff-Open-ClsFree | 3.68 | 2.12 | 0.40 | 0.54 | 0.83 | 2.92 | 1.50 | 2.54 | 46.3 | |||||||||||||||
| ConfDiff-Open-PDB | 2.90 | 1.43 | 0.38 | 0.51 | 0.82 | 2.97 | 1.57 | 2.51 | 34.1 | |||||||||||||||
| ConfDiff-Open-MD | 3.43 | 2.21 | 0.59 | 0.67 | 0.85 | 2.76 | 1.44 | 2.25 | 35.4 | |||||||||||||||
| ConfDiff-ESM-ClsFree | 4.04 | 2.84 | 0.31 | 0.43 | 0.82 | 3.82 | 1.72 | 3.06 | 37.8 | |||||||||||||||
| ConfDiff-ESM-PDB | 3.42 | 2.06 | 0.29 | 0.40 | 0.80 | 3.67 | 1.70 | 3.17 | 34.1 | |||||||||||||||
| ConfDiff-ESM-MD | 3.91 | 2.79 | 0.35 | 0.48 | 0.82 | 3.67 | 1.66 | 2.89 | 39.0 | |||||||||||||||
| Ensemble observables | Quality | |||||||||||||||||||||||
|
|
|
|
|
|
|
||||||||||||||||||
| MD iid | 0.90 | 0.80 | 0.93 | 0.56 | 0.0 | 0.1 | 3.4 | |||||||||||||||||
| MD 2.5 ns | 0.62 | 0.45 | 0.64 | 0.24 | 0.0 | 0.1 | 3.4 | |||||||||||||||||
| EigenFold | 0.36 | 0.18 | N/A | N/A | 0.7 | 9.6 | N/A | |||||||||||||||||
| MSA-depth256 | 0.30 | 0.28 | 0.33 | 0.06 | 0.0 | 0.2 | 5.9 | |||||||||||||||||
| MSA-depth64 | 0.38 | 0.27 | 0.38 | 0.12 | 0.0 | 0.2 | 8.4 | |||||||||||||||||
| MSA-depth32 | 0.39 | 0.24 | 0.36 | 0.15 | 0.1 | 0.5 | 13.0 | |||||||||||||||||
| Str2Str-ODE (t=0.1) | 0.42 | 0.17 | N/A | N/A | 0.0 | 0.1 | 13.7 | |||||||||||||||||
| Str2Str-ODE (t=0.3) | 0.41 | 0.17 | N/A | N/A | 0.0 | 0.1 | 14.8 | |||||||||||||||||
| Str2Str-SDE (t=0.1) | 0.40 | 0.13 | N/A | N/A | 1.6 | 0.2 | 23.0 | |||||||||||||||||
| Str2Str-SDE (t=0.3) | 0.35 | 0.13 | N/A | N/A | 1.5 | 0.2 | 21.4 | |||||||||||||||||
| AlphaFlow-PDB | 0.44 | 0.33 | 0.42 | 0.18 | 0.0 | 0.2 | 6.6 | |||||||||||||||||
| AlphaFlow-MD | 0.57 | 0.38 | 0.50 | 0.24 | 0.0 | 0.2 | 21.7 | |||||||||||||||||
| ESMFlow-PDB | 0.42 | 0.29 | 0.41 | 0.16 | 0.0 | 0.6 | 5.4 | |||||||||||||||||
| ESMFlow-MD | 0.51 | 0.33 | 0.47 | 0.21 | 0.0 | 0.3 | 10.9 | |||||||||||||||||
| ConfDiff-Open-PDB | 0.47 | 0.34 | 0.43 | 0.18 | 0.0 | 0.9 | 5.7 | |||||||||||||||||
| ConfDiff-Open-ClsFree | 0.54 | 0.33 | 0.47 | 0.21 | 0.0 | 1.2 | 5.7 | |||||||||||||||||
| ConfDiff-Open-MD | 0.59 | 0.36 | 0.50 | 0.24 | 0.0 | 0.8 | 6.3 | |||||||||||||||||
| ConfDiff-ESM-PDB | 0.48 | 0.31 | 0.42 | 0.18 | 0.0 | 1.6 | 3.9 | |||||||||||||||||
| ConfDiff-ESM-ClsFree | 0.54 | 0.31 | 0.47 | 0.18 | 0.0 | 1.8 | 4.3 | |||||||||||||||||
| ConfDiff-ESM-MD | 0.56 | 0.34 | 0.48 | 0.23 | 0.0 | 1.5 | 4.0 | |||||||||||||||||
4 结论和未来工作
总之,我们首次全面研究了各种蛋白质基础模型在八项不同任务中的能力,特别关注蛋白质设计和构象动力学。 我们开发了一个统一的多指标评估框架,这对于从多个方面对蛋白质基础模型进行公正评估至关重要。 基于性能结果,我们为蛋白质基础模型的开发和有效使用提供了见解和注意事项,为未来的研究提供指导。 我们重点介绍了整体评估的关键观察结果,如下所示。
4.1 关键观察
对蛋白质基础模型的有效评估需要使用正确和全面的评估指标。 以 AlphaFold2 和 ESMFold 为代表的先进折叠模型的出现,为准确评估蛋白质生成任务的质量、稳定性和精度开辟了宝贵的机会。 然而,必须承认,由于它们在复杂结构预测能力方面的当前局限性,某些任务可能仍然缺乏足够准确的评估方法。 例如,在抗体设计领域,研究人员有时会被重建指标(如氨基酸恢复 (AAR) 和均方根偏差 (RMSD))误导,这些指标与准确性相关,导致过于乐观的结果。 在这里,我们打算通过引入一种组合评估方法来解决这一挑战,该方法整合了重构和物理合理性指标。 此外,考虑到蛋白质科学问题的固有复杂性,采用多方面评估策略来捕捉蛋白质结构和功能的各个方面变得势在必行。 在 ProteinBench 中,我们旨在捕捉蛋白质结构和功能的各个方面,从而促进对蛋白质相关任务中基础模型性能的更全面理解。 此外,仅仅依靠指标是不够的。 在蛋白质生成模型的开发中,主要目标是准确地拟合训练数据的分布。 我们的评估方法超越了简单地比较指标值。 我们实施了一种更全面的评估策略,其中包括测量训练数据(包括天然蛋白质、抗体和不同长度的分子动力学构象)的相同指标。 这为蛋白质生成目标提供了高分辨率的黄金参考,允许更具上下文丰富的评估框架。
目前没有单个模型在所有蛋白质设计目标中都表现出色。 模型的选择应与预期应用仔细匹配。 在蛋白质基础模型领域,出现了两种主要方法:语言模型和几何模型。 每种方法都有其优势和局限性,这些都反映在 ProteinBench 的性能中。 我们发现语言模型在捕捉自然进化分布方面表现良好。 这在它们在天然序列恢复(逆折叠)中的高准确率和在支架进化保守基序中的高质量得到证明。 然而,语言模型在为从头骨架设计序列以及为基于序列的蛋白质设计生成新序列时,在鲁棒性方面表现出局限性。 相比之下,基于结构的模型在从头设计任务中表现出更高的鲁棒性和对结构噪声的容忍度,并且在创建具有新折叠或功能的蛋白质方面显示出更大的潜力。 这些发现强调了研究人员在选择模型时仔细考虑特定设计目标的重要性。
虽然从经典折叠模型扩展而来的生成模型已显示出对采样蛋白质构象的能力,但在多状态预测和分布预测方面仍然存在挑战。 蛋白质构象预测是评估蛋白质基础模型多模态能力和物理理解能力的一个新兴但至关重要的指标。 虽然当前模型中提出的策略可能有利于某些任务,但它们通常在其他任务中提供的改进有限。 例如,尽管使用 MD 构象数据集微调模型在 ATLAS 基准测试中显示出有希望的结果,但在 apo-holo 构象的多状态预测中几乎没有观察到改进。 此外,当前模型中多样性和质量之间的常见权衡强调了在蛋白质构象预测任务中跨准确性、多样性和质量维度进行一致评估的重要性。
4.2 局限性和未来工作
我们承认当前基准测试中存在一些局限性和改进的机会: (1) 基础模型的选择可能并不详尽。 未来迭代应纳入其他基础模型,以提供更全面的比较。 (2) 模型之间训练数据的差异目前阻碍了不同模型架构的直接比较。 未来工作可以通过标准化数据集来解决这个问题,从而允许更准确地比较架构性能。 (3) 基准测试可以扩展到包括更广泛的任务,从而进一步扩大其范围和实用性。 我们致力于不断完善和扩展 ProteinBench。 我们的愿景是使其发展成为一个动态的、不断增长的基准,以加速蛋白质建模和设计的进展。
致谢
该基准测试代表了我们研究小组的共同努力,每个成员都在各自的专业领域做出了重大贡献。 每个贡献者提供的多样化的见解和分析对塑造这项全面工作至关重要。 Q. Gu 构思并监督该项目。 F. Ye 负责协调实验和分析,同时还对逆折叠、骨架设计和部分序列设计任务进行模型评估。 Z. Zheng 负责序列设计、协同设计和基序支架中的模型评估和结果分析。 D. Xue 对抗体设计进行了模型评估和指标分析。 Y. Shen 和 L. Wang 对单态和多态预测以及构象分布预测进行了模型评估和分析。 F. Ye 发起并推动了论文的撰写,其他所有作者都做出了贡献。 我们感谢每个团队成员所展现出的奉献精神和专业知识。 他们的共同努力对于开发这个多方面的基准至关重要。
附录 A 蛋白质基础模型基准概述
在本节中,我们将全面概述现有的蛋白质基础模型基准。 表格 11 说明了这些基准的现状,揭示了范围和适用性方面的重大局限性。 大多数现有的基准都过于狭窄,主要针对特定任务的评估,而不是对蛋白质基础模型进行全面评估。
我们考察的基准可以大致分为两类:一类侧重于蛋白质设计任务,另一类评估蛋白质构象动力学。 在蛋白质设计类别中,我们观察到,尽管逆折叠在多个基准中得到了很好的体现,但其他关键方面,如骨架设计、序列设计和结构-序列协同设计,往往被忽视。 同样,在蛋白质构象动力学领域,只有少数基准涉及单态和多态预测等关键任务。
值得注意的是,我们提出的 ProteinBench 凭借其对各种任务的全面覆盖脱颖而出。 它涵盖了广泛的评估,包括蛋白质设计领域中的逆折叠、骨架设计、序列设计、结构-序列协同设计和抗体设计,以及构象动力学领域中的单态折叠和多态预测。
| Benchmark | Protein Design | Protein Conformation Prediction | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| PDB-Struct (Wang et al., 2023) | ✓ | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | ||||||||||||||||||
| Proteininvbench (Gao et al., 2024) | ✓ | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | ||||||||||||||||||
| RFDiffusion (Watson et al., 2023b) | ✘ | ✘ | ✘ | ✘ | ✓ | ✘ | ✘ | ✘ | ✘ | ||||||||||||||||||
| CASP (cas, 2022) | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | ✓ | ✘ | ✘ | ||||||||||||||||||
| CAMEO (Robin et al., 2021) | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | ✓ | ✘ | ✘ | ||||||||||||||||||
| ProteinBench | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||||||||||
附录 B 基准评估的更多细节
B.1 抗体设计
在本节中,我们将详细介绍抗体设计方法的评估,包括整体评估概念、dyMEAN 的变体、用于训练和测试的数据集,以及所有评估指标的实现。
B.1.1 评估概念
如正文所述,抗体设计最终可以简化为 CDR-H3 的设计。 因此,在本研究中,我们通过评估这些方法生成的 CDR-H3 序列来评估不同抗体设计方法的性能。 鉴于本研究的主要目标是评估各种设计模型的相对性能,而不是它们生成的抗体的体内/体外功能,我们选择直接使用其预测结构来评估设计的抗体。 这种方法基于以下几个考虑:首先,它确保明确地专注于评估设计方法本身,独立于实验约束。 其次,广泛的实验验证需要大量的时间和资源,以及准确模拟抗体真实结合结构的方法的局限性,使得体内/体外评估变得不切实际。 对设计结构的直接评估提供了一种可行且高效的策略,符合研究的目标和资源约束,同时仍然为后续的实验研究提供有价值的理论基准。
对于能够为同一抗原生成多个抗体的模型,我们使用每种方法为每个抗原生成了 64 个 CDR-H3 序列,并计算了这些不同生成的样本的平均性能。 此外,我们还计算了为单个抗原生成的不同样本的性能标准差。
B.1.2 dyMEAN 的变体
与其他方法不同,其他方法旨在接受抗体-抗原复合物的真实结构并生成缺失的 CDR-H3 区域,dyMEAN 被设置为仅接受抗原的结构和抗体非 CDR-H3 区域的序列。 因此,该模型需要同时生成CDR-H3区域并预测抗体的整体结构以及抗体和抗原之间的结合姿势。 不正确的姿势估计会严重影响CDR-H3和抗原之间的相互作用,使得dyMEAN与其他方法之间的直接比较不公平。 为了更公平地比较dyMEAN与其他方法,我们对dyMEAN进行了一些修改,提供了抗体非CDR-H3区域的真实结构和结合姿势,使dyMEAN与其他方法保持一致。 在dyMEAN-FixFR中,我们也使用Rosetta (Alford et al., 2017)重新包装侧链,与其他方法一致,以避免dyMEAN生成的侧链对评估结果的影响。 此外,我们在结构的初始化中引入了一些随机性,这使得dyMEAN-FixFR能够为相同的抗原生成多个不同的抗体。
B.1.3 数据集
为了公平比较,对所有方法进行重新训练,我们使用来自SAbDab数据集(在IMGT方案下)的抗体-抗原复合物结构数据 (Lefranc et al., 2009) 作为训练数据集。 我们收集了具有重链和轻链以及蛋白质抗原的抗原-抗体复合物。 然后我们丢弃具有相同CDR-L3和CDR-H3序列的重复数据。 其余复合物使用MMseqs2 (Steinegger & Söding, 2017) 进行聚类,以40%的序列相似度作为基于每个复合物的CDR-H3序列的阈值。 最后,我们选择不包含RAbD数据集中的复合物的聚类,并将复合物按9:1的比例(分别为1786个和193个复合物)拆分为训练集和验证集。
测试集包含从RAbD数据集中提取的55个抗体-抗原复合物。 原始RAbD数据集包含60个抗体-抗原复合物。 在本研究中,我们希望抗体设计方法的评估基于包含轻链和重链的抗体,同时抗原包含至少一条蛋白质链。 在实践中,2ghw 和 3uzq 缺少轻链,而 3h3b 缺少重链。 5d96 被排除在外,因为rabd_summary.jsonl444https://github.com/THUNLP-MT/MEAN/blob/main/summaries/rabd_summary.jsonl 中的链ID信息不正确,其中重链 J 和轻链 I 不与抗原链 A 结合。 4etq 由于 HERN 在运行此复合体时报告了错误,因此被排除在外。
B.1.4 评估指标的实现
[准确性]:
-
•
AAR:为了计算 AAR(氨基酸回收率),与现有工作类似,我们计算了生成的 CDR-H3 序列中与天然抗体匹配的残基数量。
-
•
RMSD:在计算 RMSD(均方根偏差)时,我们测量了生成的和天然抗体在 CDR-H3 区域的 CA 坐标中的 RMSD。 对于 dyMEAN 以外的方法,由于它们的任務设置提供了抗体 FR 区域和抗原的真实结合姿态,因此在计算 RMSD 时无需将生成的结构与天然结构对齐。 对于 dyMEAN,我们将 CDR-H3 两端的 2 个 FR 残基与天然抗体中的对应残基对齐,将获得的变换应用于 CDR-H3,然后计算 RMSD。
-
•
TM-score:我们仅针对 CDR-H3 区域计算 TM-score。 为此,我们将生成的 CDR-H3 部分保存为 .pdb 文件,并使用 TMalign (Zhang & Skolnick, 2005) 计算生成的 CDR-H3 和天然 CDR-H3 之间的 TM-score。
[功能性]:
-
•
结合能:结合能的计算需要蛋白质的全部原子结构,而大多数方法只生成骨架原子结构。 因此,我们首先使用 Rosetta 来填充缺失的侧链原子。 随后,我们使用 Rosetta 最小化方法优化了 CDR-H3 区域的侧链,同时保持骨架结构不变,以确保模型生成的 CDR-H3 在与抗原的结合环境中达到最低能量状态。 在最小化过程中,我们将步骤设置为 100(我们尝试使用更多步骤和重复,尽管能量进一步降低,但降低幅度非常有限,远小于不同方法之间的能量差异;但是,时间消耗显着增加)。 最小化后,我们计算了全原子结构的能量。 最后,我们使用 Rosetta 中的 InterfaceAnalyzer 计算 CDR-H3 与抗原之间的结合能。
特异性:
-
•
SeqSim:SeqSim 定义为生成的序列中任意序列对之间的平均相似度。 首先,我们介绍相似度的定义和实现。 两个序列之间的相似度定义为对齐后匹配氨基酸的百分比,占对齐长度的比例(因此,该指标受两个序列之间长度差距的影响)。 鉴于我们的目标是计算匹配的数量而不是匹配得分,并且 CDR-H3 的两端固定到 FR3 和 FR4,我们需要一种对齐方法,该方法:(1)对匹配分配 1 分,对间隙和不匹配分配 0 分;(2)不会在 CDR-H3 的两端引入间隙。 我们使用 Biopython 中的 PairwiseAligner (Cock 等人,2009) 进行序列比对,将 match_score 设置为 1,所有其他得分设置为 0,end_gap_score 设置为 -inf,以便对齐过程满足我们的要求。 对于仅生成一个抗体/抗原的方法,我们直接计算 55 个生成的 CDR-H3 序列中 SeqSim 的平均值作为 SeqSim-outer。 对于生成多个抗体的方法,我们计算为两个抗原生成的两个序列集之间的平均 SeqSim 作为 SeqSim-outer,并计算每个集合内的平均 SeqSim 作为 SeqSim-inner。 计算 SeqSim-outer 和 SeqSim-inner 的公式如下:
SeqSim-outer (1) SeqSim-inner (2) 其中 表示测试集中抗原的数量(=55 本研究), 表示为每个抗原生成的样本数量(=64 本研究), 表示为第 个抗原生成的第 个 CDR-H3 序列。
-
•
PHR:PHR 是生成的 CDR-H3 序列中疏水残基的比例。 尽管 PHR 和 SeqSim 都用于表示抗体设计方法的特异性,但它们关注的是不同的方面。 因此,同一种方法在这些指标上可能表现出不同的趋势(SeqSim 可以理解为对方法特异性的评估,而 PHR 是对生成的抗体特异性的评估。 当 SeqSim 表现不佳时,PHR 的性能意义有限)。 例如,AbDPO 实现了较高的 SeqSim-outer,但在 PHR 中表现不佳。 这表明 AbDPO 可以专门为不同的抗原设计抗体,但这些抗体包含许多疏水残基,导致与多种蛋白质发生潜在的非特异性相互作用。
[理性]:
-
•
CN-Score: 为了评估生成的抗体肽键长度与天然抗体肽键长度的一致性,我们使用天然抗体CDR-H3区域中发现的肽键长度拟合核密度估计(KDE)函数。 生成的肽键长度的密度,即CN-Score,用来表示一致性。 对于生成的肽键长度小于最小天然肽键长度或大于最大天然肽键长度,其密度定义为0。 生成的抗体的最终CN-Score定义为所有肽键长度的平均密度。 值得注意的是,肽键长度的变化非常小,导致天然肽键长度的分布非常窄。 当生成的肽键长度略微偏离平均长度(1.3310)时,其在KDE函数中的密度将急剧下降,这解释了为什么所有方法在CN-Score上与天然抗体相比都存在显著差异。
-
•
碰撞: 虽然蛋白质内部的原子碰撞主要发生在侧链之间,但大多数方法不会生成残基的侧链。 使用填充方法来完成侧链可以通过广泛的搜索始终找到碰撞最少的侧链构象。 因此,我们评估了生成结构中的潜在碰撞水平,而不是具体的碰撞数量。 为此,我们计算了两个残基之间的CA距离;当两个残基之间的CA-CA距离小于共价键连接的残基中通常发现的最小CA-CA距离(3.6574,来自RAbD数据集CDR-H3区域的CA-CA距离统计)时,我们认为这两个残基存在潜在的碰撞。 然后,我们计算距离低于此阈值的残基对的数量,以衡量生成结构中的碰撞水平。 Clashes-inner和Clashes-outer之间的区别是:Clashes-inner衡量生成CDR-H3结构内部的碰撞水平,而Clashes-outer衡量生成CDR-H3结构与其他组件(包括抗原、重链FR区域和抗体的轻链)之间的碰撞水平。
-
•
SeqNat: 为了衡量设计的CDR-H3序列与天然序列的接近程度,我们使用了AntiBERTy模型预测的pLL。 我们将整个重链序列输入模型,这意味着AntiBERTy基于抗体的整个重链进行预测,但与AntiBERTy中的标准程序不同,pLL计算区域仅在CDR-H3区域内(标准程序在整个输入序列上计算pLL)。
-
•
总能量: 在计算总能量之前,我们对设计的CDR-H3区域进行了与功能部分描述相同的能量优化过程。 然后,我们使用 Rosetta 的全原子评分函数,并使用 REF15 (Alford 等人,2017) 中的默认权重来计算 CDR-H3 区域中每个残基的总能量。 CDR-H3 区域的总能量定义为其所有残基的总能量之和。
-
•
scRMSD:在此度量中,我们使用 IgFold 来预测生成的序列的结构。 IgFold 基于抗体轻链和重链的序列对来预测结构(尽管我们评估的区域只存在于重链中,并且 IgFold 也支持单链输入,但我们发现输入两条链可以获得更高的准确率)。 抗体非 CDR-H3 区域的真实结构也作为模板提供,以获得初始的预测结构。 然后,我们使用 Kabsch 算法将重链的非 CDR-H3 区域与真实结构对齐,并将得到的变换应用于预测的 CDR-H3 结构。 这将预测的 CDR-H3 结构与原始复合体对齐。 此时,预测的 CDR-H3 结构与 RAbD 数据集中真实结构的 CA-RMSD 为 1.95。 IgFold 预测的结构与抗原无关,并且由于抗体在与抗原结合后在结合界面经历构象变化,我们使用 Rosetta 在抗原存在的情况下对预测的 CDR-H3 进行弛豫。 弛豫涉及主链和侧链结构的变化。 具体而言,我们对 IgFold 预测的每个结构重复弛豫运行五次,每次 200 步,并选择能量最低的结构作为最终预测结构。 在此阶段,与真实结构的 CA-RMSD 降至 1.77。 然后,我们计算了预测结构与模型生成的骨架 CA 坐标之间的 CA 坐标的 RMSD,称为 scRMSD。
B.2 蛋白质构象预测
本节将详细介绍 蛋白质构象预测 基准测试中使用的数据集、评估指标和模型实现。
B.2.1 数据集
-
•
CAMEO2022 包含从 2022 年 8 月到 10 月从 CAMEO 目标中收集的 183 条单蛋白质链,其序列长度小于 750 个氨基酸,遵循 Jing 等人 (2023)。 使用自定义脚本从 RCSB 蛋白质数据库 (https://www.rcsb.org/, Berman 等人 (2000)) 中可用的 mmCIF 文件中提取蛋白质序列和结构。 其中一种蛋白质 (PDB ID: 8AHP, 链 A) 已被新的 PDB 条目 8QCW 取代,我们已用更新的记录替换了这条链。
-
•
Apo-holo 包含 91 条由 Saldaño 等人 (2022) 策划的单链蛋白质。 使用与 CAMEO2022 中相同的流程提取了 apo 和 holo 两种构象的蛋白质序列和结构。 遵循 Jing 等人 (2023),我们使用 apo 结构的序列作为模型推断的主要序列。
-
•
BPTI 是一种包含 58 个氨基酸的蛋白质,其动力学已通过长时间 MD 模拟 (Shaw 等人, 2010) 进行了广泛研究。 我们使用 MD 研究中确定的聚类中心的结构作为评估的参考结构。
-
•
ATLAS 是一个最近发布的数据集,包含 1,390 种不同的单链蛋白质的 100 ns MD 模拟三联体 (Vander Meersche 等人, 2024)。 在这项工作中,我们使用了一个子集,其中包含 82 种蛋白质,其 PDB 条目是在 2019 年 5 月 1 日之后提交的,遵循 Jing 等人 (2024)。 从 ATLAS 数据库 555https://www.dsimb.inserm.fr/ATLAS/index.html 下载了“仅蛋白质”轨迹以进行评估。
B.2.2 评估指标
[准确性]
我们通过将生成的构象与参考结构进行比较来评估准确性。 具体来说,TMscore、RMSD、GDT-TS 是使用 TMscore Zhang & Skolnick (2004) 计算的,该值来自 https://zhanggroup.org/TM-score/。 我们使用 -seq 选项在结构比对之前对齐序列。 lDDT 分数使用原始实现 Mariani 等人 (2013) 计算,可从 https://swissmodel.expasy.org/lddt/downloads/ 获取。
在多状态预测中,我们通过生成的样本中最佳的准确率来评估预测特定状态的准确率。 例如,apo-TM) 是样本 (基准中 N=20) 和参考 apo 结构之间的最高 TM 分数。 遵循 Jing 等人 (2023),我们使用“集成准确率”评估预测多个状态的总体能力,这是每个状态的准确率的平均值,由 TM 分数或 RMSD 衡量。
对于分布预测任务中的准确度指标(灵活性、分布准确度、集成可观察量),我们遵循实现 666https://github.com/bjing2016/alphaflow/blob/master/scripts/analyze_ensembles.py 的 Jing 等人 (2024)。 以下是这些指标的概述:灵活性(或 Jing 等人 (2024) 中预测的灵活性)反映了模型预测蛋白质(成对 RMSD)或原子(RMSF)多样性的准确程度。 这是通过模型生成样本的多样性度量(简称 样本分布)和参考 MD 样本(简称 参考分布)之间的皮尔逊相关系数 来衡量的。 分布准确度 直接比较样本和参考分布之间的相似性。 RMWD 是对齐坐标分布的根均值 Wasserstein 距离,建模为多元高斯分布。 在此基准测试中,我们仅报告总距离,不进行平移和方差分解。 此外,我们评估了前两个 PCA 维度(对齐坐标)中构象分布的 Wasserstein-2 距离,以及样本和参考分布的 PCA 分量之间的余弦相似度。 最后,集成可观察量 包括比较样本和参考分布中特定可观察量(即感兴趣的属性)的指标,特别是残基-残基接触(弱或瞬时)和残基暴露(例如,与溶剂接触的表面残基)。 Jaccard 相似性和互信息 (MI) 用于比较这些可观察量。
[多样性]
多样性通过蛋白质生成样本之间的平均成对结构相似性来评估,使用 TM 分数或 RMSD 测量。 为了减少计算时间,我们随机抽取 100 对结构用于此计算。
[质量]
生成的构象结构的质量通过三个主链结构违规来评估:CA-clash %,CA-break % 和 PepBond-break %。
-
•
CA-clash % 是α-碳原子之间发生碰撞的比率。 如果一对α-碳原子之间的距离小于 3.0 Å,则确定为碰撞,类似于Lu 等人 (2024)。 并且 CA-clash % 计算为
-
•
CA-break % 是两个连接残基距离过远导致潜在键断裂的比率。 如果两个连接残基之间的距离大于 4.2 Å,我们确定为断裂,并且 CA-break % 计算为
-
•
PepBond-break % 特别评估连接残基之间潜在的肽键 (C-N) 断裂,提供了比 CA-break % 更严格的关于残基间断裂的指标。 我们使用 1.4 Å 的最大肽长度阈值来确定链断裂,如 Biopython 包中777https://biopython.org/docs/dev/api/Bio.PDB.internal_coords.html#Bio.PDB.internal_coords.IC_Chain。 同样,PepBond-break % 计算为
B.2.3 模型实现
-
•
AlphaFold2 (Jumper 等人,2021):我们使用 ColabFold 实现Mirdita 等人 (2022) 进行 AlphaFold2 推理,输入 MSA 使用 Colab 管道获得。 所有五个模型(具有 pTM)用于预测五个候选结构,并选择 pLDDT 置信度得分最高的结构进行性能评估。 所有模型都以默认设置运行,并且没有提供模板。
-
•
OpenFold (Ahdritz 等人,2022):我们使用openfold v2.0.0 进行推理,并使用他们的预训练 OpenFold 权重(具有 pTM)。 与用于 AlphaFold2 的相同 MSA 作为输入提供。 由于只有一个与模型配置 model_3_ptm 相对应的检查点 (finetuning_no_templ_ptm_1) 可用,我们使用三个随机种子生成了三个结构,并总共进行了 5 次预测。 选择具有最高 pLDDT 分数的结构进行性能评估。 推断时未提供模板。
-
•
ESMFold (Lin 等人,2023):我们使用公共 ESM 存储库进行推理,模型为 esm.pretrained.esmfold_v1()。 由于 EMSFold 预测是确定性的,因此我们只为每个蛋白质生成了一个结构用于性能评估。
-
•
RoseTTAFold2 (Baek 等人,2023):我们遵循他们的官方存储库和推理说明,使用与 AlphaFold2 和 OpenFold 相同的 MSA。 未提供任何模板。 每个蛋白质只预测了一个结构用于性能评估。
-
•
EigenFold (Jing 等人,2023):我们遵循作者提供的官方存储库、权重和推理设置。 在 蛋白质折叠 任务中,我们为每个蛋白质采样了 5 个结构,并选择 ELBO 估计值最高的结构进行性能评估。 由于 EigenFold 无法预测包含未知氨基酸(标记为“X”)的序列,因此我们像原始实现那样删除了输入序列中的“X”。 这种调整可能会导致由于推理使用略微不同的序列而产生一些性能差异。
-
•
MSA 子采样 (Del Alamo 等人,2022):我们使用 openfold v2.0.0 包实现了 MSA 子采样,通过调整两个配置参数 max_msa_clusters 和 max_extra_msa,遵循 Del Alamo 等人 (2022)。 具体来说,我们将 max_extra_msa 称为 MSA 深度,并将 max_msa_clusters 设置为深度的一半,同时将其他 OpenFold 设置保持为默认值。 使用与 AlphaFold2 中相同的 ColabFold 管道获取原始 MSA。
-
•
Str2Str (Lu 等人,2024):我们遵循 Str2Str 的官方实现,并使用 OpenFold 预测的结构作为初始结构。 通过从 值中进行均匀采样来收集集成结果。 对于 BPTI,我们使用作者推荐的噪声调度,最大正向时间为 。 对于 apo-holo 和 ATLAS 数据集,我们对 SDE 和 ODE 模型都尝试了 和 。
-
•
AlphaFlow/ESMFlow (Jing et al., 2024): 我们使用了官方存储库和发布的模型权重进行推理。 AlphaFlow 模型的 MSAs 通过 ColabFold 的管道获得。
-
•
ConfDiff (Wang et al., 2024c): 我们遵循作者的实现并使用发布的权重进行推理。 在此基准测试中,我们对 ConfDiff-Open 和 ConfDiff-ESM 模型都使用了 recycle3 表示。 能量和力引导模型是特定于数据集的,并且仅适用于具有 ESMFold 表示的 BPTI 数据集。
参考文献
- cas (2022) Abstract book of the 15th critical assessment of structure prediction. 2022. URL https://predictioncenter.org/casp15/doc/CASP15Abstracts.pdf.
- Abramson et al. (2024) Josh Abramson, Jonas Adler, Jack Dunger, Richard Evans, Tim Green, Alexander Pritzel, Olaf Ronneberger, Lindsay Willmore, Andrew J Ballard, Joshua Bambrick, et al. Accurate structure prediction of biomolecular interactions with alphafold 3. Nature, pp. 1–3, 2024.
- Adolf-Bryfogle et al. (2018) Jared Adolf-Bryfogle, Oleks Kalyuzhniy, Michael Kubitz, Brian D Weitzner, Xiaozhen Hu, Yumiko Adachi, William R Schief, and Roland L Dunbrack Jr. Rosettaantibodydesign (rabd): A general framework for computational antibody design. PLoS computational biology, 14(4):e1006112, 2018.
- Ahdritz et al. (2022) Gustaf Ahdritz, Nazim Bouatta, Christina Floristean, Sachin Kadyan, Qinghui Xia, William Gerecke, Timothy J O’Donnell, Daniel Berenberg, Ian Fisk, Niccolò Zanichelli, Bo Zhang, Arkadiusz Nowaczynski, Bei Wang, Marta M Stepniewska-Dziubinska, Shang Zhang, Adegoke Ojewole, Murat Efe Guney, Stella Biderman, Andrew M Watkins, Stephen Ra, Pablo Ribalta Lorenzo, Lucas Nivon, Brian Weitzner, Yih-En Andrew Ban, Peter K Sorger, Emad Mostaque, Zhao Zhang, Richard Bonneau, and Mohammed AlQuraishi. OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. bioRxiv, 2022. doi: 10.1101/2022.11.20.517210. URL https://www.biorxiv.org/content/10.1101/2022.11.20.517210.
- Alamdari et al. (2023) Sarah Alamdari, Nitya Thakkar, Rianne van den Berg, Alex X Lu, Nicolo Fusi, Ava P Amini, and Kevin K Yang. Protein generation with evolutionary diffusion: sequence is all you need. In Machine Learning for Structural Biology Workshop, NeurIPS 2023, 2023.
- Alford et al. (2017) Rebecca F Alford, Andrew Leaver-Fay, Jeliazko R Jeliazkov, Matthew J O’Meara, Frank P DiMaio, Hahnbeom Park, Maxim V Shapovalov, P Douglas Renfrew, Vikram K Mulligan, Kalli Kappel, et al. The rosetta all-atom energy function for macromolecular modeling and design. Journal of chemical theory and computation, 13(6):3031–3048, 2017.
- Alley et al. (2019) Ethan C. Alley, Grigory Khimulya, Surojit Biswas, Mohammed AlQuraishi, and George M. Church. Unified rational protein engineering with sequence-based deep representation learning. Nature Methods, 16:1315–1322, 12 2019. ISSN 1548-7091. doi: 10.1038/s41592-019-0598-1.
- Baek et al. (2023) Minkyung Baek, Ivan Anishchenko, Ian R Humphreys, Qian Cong, David Baker, and Frank DiMaio. Efficient and accurate prediction of protein structure using rosettafold2. BioRxiv, pp. 2023–05, 2023.
- Berman et al. (2000) Helen M Berman, John Westbrook, Zukang Feng, Gary Gilliland, Talapady N Bhat, Helge Weissig, Ilya N Shindyalov, and Philip E Bourne. The protein data bank. Nucleic acids research, 28(1):235–242, 2000.
- Bose et al. (2023) Avishek Joey Bose, Tara Akhound-Sadegh, Kilian Fatras, Guillaume Huguet, Jarrid Rector-Brooks, Cheng-Hao Liu, Andrei Cristian Nica, Maksym Korablyov, Michael Bronstein, and Alexander Tong. Se (3)-stochastic flow matching for protein backbone generation. arXiv preprint arXiv:2310.02391, 2023.
- (11) Andrew Campbell, Jason Yim, Regina Barzilay, Tom Rainforth, and Tommi Jaakkola. Generative flows on discrete state-spaces: Enabling multimodal flows with applications to protein co-design. In Forty-first International Conference on Machine Learning.
- Chu et al. (2024) Alexander E Chu, Jinho Kim, Lucy Cheng, Gina El Nesr, Minkai Xu, Richard W Shuai, and Po-Ssu Huang. An all-atom protein generative model. Proceedings of the National Academy of Sciences, 121(27):e2311500121, 2024.
- Cock et al. (2009) Peter JA Cock, Tiago Antao, Jeffrey T Chang, Brad A Chapman, Cymon J Cox, Andrew Dalke, Iddo Friedberg, Thomas Hamelryck, Frank Kauff, Bartek Wilczynski, et al. Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11):1422, 2009.
- Dauparas et al. (2022) J. Dauparas, I. Anishchenko, N. Bennett, H. Bai, R. J. Ragotte, L. F. Milles, B. I. M. Wicky, A. Courbet, R. J. de Haas, N. Bethel, P. J. Y. Leung, T. F. Huddy, S. Pellock, D. Tischer, F. Chan, B. Koepnick, H. Nguyen, A. Kang, B. Sankaran, A. K. Bera, N. P. King, and D. Baker. Robust deep learning–based protein sequence design using proteinmpnn. Science, 378:49–56, 10 2022. ISSN 0036-8075. doi: 10.1126/science.add2187.
- Del Alamo et al. (2022) Diego Del Alamo, Davide Sala, Hassane S Mchaourab, and Jens Meiler. Sampling alternative conformational states of transporters and receptors with alphafold2. Elife, 11:e75751, 2022.
- Dunbar et al. (2013) James Dunbar, Konrad Krawczyk, Jinwoo Leem, Terry Baker, Angelika Fuchs, Guy Georges, Jiye Shi, and Charlotte M. Deane. SAbDab: the structural antibody database. Nucleic Acids Research, 42(D1):D1140–D1146, 11 2013. ISSN 0305-1048. doi: 10.1093/nar/gkt1043. URL https://doi.org/10.1093/nar/gkt1043.
- Gao et al. (2022) Zhangyang Gao, Cheng Tan, Pablo Chacón, and Stan Z Li. Pifold: Toward effective and efficient protein inverse folding. arXiv preprint arXiv:2209.12643, 2022.
- Gao et al. (2024) Zhangyang Gao, Cheng Tan, Yijie Zhang, Xingran Chen, Lirong Wu, and Stan Z Li. Proteininvbench: Benchmarking protein inverse folding on diverse tasks, models, and metrics. Advances in Neural Information Processing Systems, 36, 2024.
- Hayes et al. (2024) Tomas Hayes, Roshan Rao, Halil Akin, Nicholas J Sofroniew, Deniz Oktay, Zeming Lin, Robert Verkuil, Vincent Q Tran, Jonathan Deaton, Marius Wiggert, et al. Simulating 500 million years of evolution with a language model. bioRxiv, pp. 2024–07, 2024.
- Hie et al. (2024) Brian L. Hie, Varun R. Shanker, Duo Xu, Theodora U. J. Bruun, Payton A. Weidenbacher, Shaogeng Tang, Wesley Wu, John E. Pak, and Peter S. Kim. Efficient evolution of human antibodies from general protein language models. Nature Biotechnology, 42:275–283, 2 2024. ISSN 1087-0156. doi: 10.1038/s41587-023-01763-2.
- Hsu et al. (2022) Chloe Hsu, Robert Verkuil, Jason Liu, Zeming Lin, Brian Hie, om Sercu, Adam Lerer, and Alexander Rives. Learning inverse folding from millions of predicted structures. Proceedings of the 39th International Conference on Machine Learning, 162, 2022. doi: 10.1101/2022.04.10.487779.
- Ingraham et al. (2023) John B Ingraham, Max Baranov, Zak Costello, Karl W Barber, Wujie Wang, Ahmed Ismail, Vincent Frappier, Dana M Lord, Christopher Ng-Thow-Hing, Erik R Van Vlack, et al. Illuminating protein space with a programmable generative model. Nature, 623(7989):1070–1078, 2023.
- Jin et al. (2022) Wengong Jin, Regina Barzilay, and Tommi Jaakkola. Antibody-antigen docking and design via hierarchical structure refinement. In International Conference on Machine Learning, pp. 10217–10227. PMLR, 2022.
- Jing et al. (2020) Bowen Jing, Stephan Eismann, Patricia Suriana, Raphael John Lamarre Townshend, and Ron Dror. Learning from protein structure with geometric vector perceptrons. In International Conference on Learning Representations, 2020.
- Jing et al. (2023) Bowen Jing, Ezra Erives, Peter Pao-Huang, Gabriele Corso, Bonnie Berger, and Tommi S Jaakkola. Eigenfold: Generative protein structure prediction with diffusion models. In ICLR 2023-Machine Learning for Drug Discovery workshop, 2023.
- Jing et al. (2024) Bowen Jing, Bonnie Berger, and Tommi Jaakkola. Alphafold meets flow matching for generating protein ensembles. In Forty-first International Conference on Machine Learning, 2024.
- Jumper et al. (2021) John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, Alex Bridgland, Clemens Meyer, Simon A. A. Kohl, Andrew J. Ballard, Andrew Cowie, Bernardino Romera-Paredes, Stanislav Nikolov, Rishub Jain, Jonas Adler, Trevor Back, Stig Petersen, David Reiman, Ellen Clancy, Michal Zielinski, Martin Steinegger, Michalina Pacholska, Tamas Berghammer, Sebastian Bodenstein, David Silver, Oriol Vinyals, Andrew W. Senior, Koray Kavukcuoglu, Pushmeet Kohli, and Demis Hassabis. Highly accurate protein structure prediction with alphafold. Nature, 596:583–589, 8 2021. ISSN 0028-0836. doi: 10.1038/s41586-021-03819-2.
- Kong et al. (2022) Xiangzhe Kong, Wenbing Huang, and Yang Liu. Conditional antibody design as 3d equivariant graph translation. In The Eleventh International Conference on Learning Representations. ICLR, 2022.
- Kong et al. (2023) Xiangzhe Kong, Wenbing Huang, and Yang Liu. End-to-end full-atom antibody design. In Proceedings of the 40th International Conference on Machine Learning, pp. 17409–17429, 2023.
- Krishna et al. (2024) Rohith Krishna, Jue Wang, Woody Ahern, Pascal Sturmfels, Preetham Venkatesh, Indrek Kalvet, Gyu Rie Lee, Felix S. Morey-Burrows, Ivan Anishchenko, Ian R. Humphreys, Ryan McHugh, Dionne Vafeados, Xinting Li, George A. Sutherland, Andrew Hitchcock, C. Neil Hunter, Alex Kang, Evans Brackenbrough, Asim K. Bera, Minkyung Baek, Frank DiMaio, and David Baker. Generalized biomolecular modeling and design with rosettafold all-atom. Science, 384, 4 2024. ISSN 0036-8075. doi: 10.1126/science.adl2528.
- Kuhlman & Bradley (2019) Brian Kuhlman and Philip Bradley. Advances in protein structure prediction and design. Nature Reviews Molecular Cell Biology, 20:681–697, 11 2019. ISSN 1471-0072. doi: 10.1038/s41580-019-0163-x.
- Lefranc et al. (2009) Marie-Paule Lefranc, Veronique Giudicelli, Chantal Ginestoux, Joumana Jabado-Michaloud, Geraldine Folch, Fatena Bellahcene, Yan Wu, Elodie Gemrot, Xavier Brochet, Jeroˆme Lane, et al. Imgt®, the international immunogenetics information system®. Nucleic acids research, 37(suppl_1):D1006–D1012, 2009.
- Lin & AlQuraishi (2023) Yeqing Lin and Mohammed AlQuraishi. Generating novel, designable, and diverse protein structures by equivariantly diffusing oriented residue clouds. arXiv preprint arXiv:2301.12485, 2023.
- Lin et al. (2023) Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, Allan dos Santos Costa, Maryam Fazel-Zarandi, Tom Sercu, Salvatore Candido, and Alexander Rives. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science, 379:1123–1130, 3 2023. ISSN 0036-8075. doi: 10.1126/science.ade2574.
- Lisanza et al. (2023) Sidney Lyayuga Lisanza, Jake Merle Gershon, Sam Tipps, Lucas Arnoldt, Samuel Hendel, Jeremiah Nelson Sims, Xinting Li, and David Baker. Joint generation of protein sequence and structure with rosettafold sequence space diffusion. bioRxiv, pp. 2023–05, 2023.
- Liu et al. (2023) Haiyan Liu, Yufeng Liu, and Linghui Chen. Diffusion in a quantized vector space generates non-idealized protein structures and predicts conformational distributions. bioRxiv, pp. 2023–11, 2023.
- Lu et al. (2024) Jiarui Lu, Bozitao Zhong, Zuobai Zhang, and Jian Tang. Str2str: A score-based framework for zero-shot protein conformation sampling. In The Twelfth International Conference on Learning Representations, 2024.
- Luo et al. (2022) Shitong Luo, Yufeng Su, Xingang Peng, Sheng Wang, Jian Peng, and Jianzhu Ma. Antigen-specific antibody design and optimization with diffusion-based generative models for protein structures. Advances in Neural Information Processing Systems, 35:9754–9767, 2022.
- Madani et al. (2023) Ali Madani, Ben Krause, Eric R. Greene, Subu Subramanian, Benjamin P. Mohr, James M. Holton, Jose Luis Olmos, Caiming Xiong, Zachary Z. Sun, Richard Socher, James S. Fraser, and Nikhil Naik. Large language models generate functional protein sequences across diverse families. Nature Biotechnology, 41:1099–1106, 8 2023. ISSN 1087-0156. doi: 10.1038/s41587-022-01618-2.
- Mariani et al. (2013) Valerio Mariani, Marco Biasini, Alessandro Barbato, and Torsten Schwede. lddt: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics, 29(21):2722–2728, 2013.
- Mirdita et al. (2022) Milot Mirdita, Konstantin Schütze, Yoshitaka Moriwaki, Lim Heo, Sergey Ovchinnikov, and Martin Steinegger. Colabfold: making protein folding accessible to all. Nature methods, 19(6):679–682, 2022.
- Nijkamp et al. (2023) Erik Nijkamp, Jeffrey A. Ruffolo, Eli N. Weinstein, Nikhil Naik, and Ali Madani. Progen2: Exploring the boundaries of protein language models. Cell Systems, 14:968–978.e3, 11 2023. ISSN 24054712. doi: 10.1016/j.cels.2023.10.002.
- Notin et al. (2024) Pascal Notin, Aaron Kollasch, Daniel Ritter, Lood Van Niekerk, Steffanie Paul, Han Spinner, Nathan Rollins, Ada Shaw, Rose Orenbuch, Ruben Weitzman, et al. Proteingym: Large-scale benchmarks for protein fitness prediction and design. Advances in Neural Information Processing Systems, 36, 2024.
- Ren et al. (2024) Milong Ren, Chungong Yu, Dongbo Bu, and Haicang Zhang. Accurate and robust protein sequence design with carbondesign. Nature Machine Intelligence, 6(5):536–547, 2024.
- Rives et al. (2021) Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C. Lawrence Zitnick, Jerry Ma, and Rob Fergus. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences, 118, 4 2021. ISSN 0027-8424. doi: 10.1073/pnas.2016239118.
- Robin et al. (2021) Xavier Robin, Juergen Haas, Rafal Gumienny, Anna Smolinski, Gerardo Tauriello, and Torsten Schwede. Continuous automated model evaluation (cameo)—perspectives on the future of fully automated evaluation of structure prediction methods. Proteins: Structure, Function, and Bioinformatics, 89:1977–1986, 12 2021. ISSN 0887-3585. doi: 10.1002/prot.26213.
- Ruffolo et al. (2021) Jeffrey A Ruffolo, Jeffrey J Gray, and Jeremias Sulam. Deciphering antibody affinity maturation with language models and weakly supervised learning. In Machine Learning for Structural Biology Workshop, NeurIPS 2021., 2021.
- Ruffolo et al. (2023) Jeffrey A Ruffolo, Lee-Shin Chu, Sai Pooja Mahajan, and Jeffrey J Gray. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Nature communications, 14(1):2389, 2023.
- Saldaño et al. (2022) Tadeo Saldaño, Nahuel Escobedo, Julia Marchetti, Diego Javier Zea, Juan Mac Donagh, Ana Julia Velez Rueda, Eduardo Gonik, Agustina García Melani, Julieta Novomisky Nechcoff, Martín N Salas, et al. Impact of protein conformational diversity on alphafold predictions. Bioinformatics, 38(10):2742–2748, 2022.
- Shaw et al. (2010) David E Shaw, Paul Maragakis, Kresten Lindorff-Larsen, Stefano Piana, Ron O Dror, Michael P Eastwood, Joseph A Bank, John M Jumper, John K Salmon, Yibing Shan, et al. Atomic-level characterization of the structural dynamics of proteins. Science, 330(6002):341–346, 2010.
- Shin et al. (2021) Jung-Eun Shin, Adam J. Riesselman, Aaron W. Kollasch, Conor McMahon, Elana Simon, Chris Sander, Aashish Manglik, Andrew C. Kruse, and Debora S. Marks. Protein design and variant prediction using autoregressive generative models. Nature Communications, 12:2403, 4 2021. ISSN 2041-1723. doi: 10.1038/s41467-021-22732-w.
- Steinegger & Söding (2017) Martin Steinegger and Johannes Söding. Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature biotechnology, 35(11):1026–1028, 2017.
- Trippe et al. (2022) Brian L Trippe, Jason Yim, Doug Tischer, David Baker, Tamara Broderick, Regina Barzilay, and Tommi Jaakkola. Diffusion probabilistic modeling of protein backbones in 3d for the motif-scaffolding problem. arXiv preprint arXiv:2206.04119, 2022.
- van Kempen et al. (2022) Michel van Kempen, Stephanie S Kim, Charlotte Tumescheit, Milot Mirdita, Cameron LM Gilchrist, Johannes Söding, and Martin Steinegger. Foldseek: fast and accurate protein structure search. Biorxiv, pp. 2022–02, 2022.
- Vander Meersche et al. (2024) Yann Vander Meersche, Gabriel Cretin, Aria Gheeraert, Jean-Christophe Gelly, and Tatiana Galochkina. Atlas: protein flexibility description from atomistic molecular dynamics simulations. Nucleic acids research, 52(D1):D384–D392, 2024.
- Varadi et al. (2022) Mihaly Varadi, Stephen Anyango, Mandar Deshpande, Sreenath Nair, Cindy Natassia, Galabina Yordanova, David Yuan, Oana Stroe, Gemma Wood, Agata Laydon, et al. Alphafold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic acids research, 50(D1):D439–D444, 2022.
- Verkuil et al. (2022) Robert Verkuil, Ori Kabeli, Yilun Du, Basile IM Wicky, Lukas F Milles, Justas Dauparas, David Baker, Sergey Ovchinnikov, Tom Sercu, and Alexander Rives. Language models generalize beyond natural proteins. BioRxiv, pp. 2022–12, 2022.
- Wang et al. (2024a) Chentong Wang, Yannan Qu, Zhangzhi Peng, Yukai Wang, Hongli Zhu, Dachuan Chen, and Longxing Cao. Proteus: exploring protein structure generation for enhanced designability and efficiency. bioRxiv, pp. 2024–02, 2024a.
- Wang et al. (2023) Chuanrui Wang, Bozitao Zhong, Zuobai Zhang, Narendra Chaudhary, Sanchit Misra, and Jian Tang. Pdb-struct: A comprehensive benchmark for structure-based protein design. arXiv preprint arXiv:2312.00080, 2023.
- Wang et al. (2024b) Xinyou Wang, Zaixiang Zheng, Fei Ye, Dongyu Xue, Shujian Huang, and Quanquan Gu. Diffusion language models are versatile protein learners. In International conference on machine learning, 2024b.
- Wang et al. (2024c) Yan Wang, Lihao Wang, Yuning Shen, Yiqun Wang, Huizhuo Yuan, Yue Wu, and Quanquan Gu. Protein conformation generation via force-guided se (3) diffusion models. In Forty-first International Conference on Machine Learning, 2024c.
- Watson et al. (2023a) Joseph L. Watson, David Juergens, Nathaniel R. Bennett, Brian L. Trippe, Jason Yim, Helen E. Eisenach, Woody Ahern, Andrew J. Borst, Robert J. Ragotte, Lukas F. Milles, Basile I. M. Wicky, Nikita Hanikel, Samuel J. Pellock, Alexis Courbet, William Sheffler, Jue Wang, Preetham Venkatesh, Isaac Sappington, Susana Vázquez Torres, Anna Lauko, Valentin De Bortoli, Emile Mathieu, Sergey Ovchinnikov, Regina Barzilay, Tommi S. Jaakkola, Frank DiMaio, Minkyung Baek, and David Baker. De novo design of protein structure and function with rfdiffusion. Nature, 620:1089–1100, 8 2023a. ISSN 0028-0836. doi: 10.1038/s41586-023-06415-8.
- Watson et al. (2023b) Joseph L Watson, David Juergens, Nathaniel R Bennett, Brian L Trippe, Jason Yim, Helen E Eisenach, Woody Ahern, Andrew J Borst, Robert J Ragotte, Lukas F Milles, et al. De novo design of protein structure and function with rfdiffusion. Nature, 620(7976):1089–1100, 2023b.
- Wayment-Steele et al. (2024) Hannah K Wayment-Steele, Adedolapo Ojoawo, Renee Otten, Julia M Apitz, Warintra Pitsawong, Marc Hömberger, Sergey Ovchinnikov, Lucy Colwell, and Dorothee Kern. Predicting multiple conformations via sequence clustering and alphafold2. Nature, 625(7996):832–839, 2024.
- Wu et al. (2024a) Kevin E Wu, Kevin K Yang, Rianne van den Berg, Sarah Alamdari, James Y Zou, Alex X Lu, and Ava P Amini. Protein structure generation via folding diffusion. Nature communications, 15(1):1059, 2024a.
- Wu et al. (2024b) Luhuan Wu, Brian Trippe, Christian Naesseth, David Blei, and John P Cunningham. Practical and asymptotically exact conditional sampling in diffusion models. Advances in Neural Information Processing Systems, 36, 2024b.
- Wu et al. (2022) Ruidong Wu, Fan Ding, Rui Wang, Rui Shen, Xiwen Zhang, Shitong Luo, Chenpeng Su, Zuofan Wu, Qi Xie, Bonnie Berger, et al. High-resolution de novo structure prediction from primary sequence. BioRxiv, pp. 2022–07, 2022.
- Yim et al. (2023) Jason Yim, Andrew Campbell, Andrew YK Foong, Michael Gastegger, José Jiménez-Luna, Sarah Lewis, Victor Garcia Satorras, Bastiaan S Veeling, Regina Barzilay, Tommi Jaakkola, et al. Fast protein backbone generation with se (3) flow matching. arXiv preprint arXiv:2310.05297, 2023.
- Yim et al. (2024) Jason Yim, Andrew Campbell, Emile Mathieu, Andrew YK Foong, Michael Gastegger, José Jiménez-Luna, Sarah Lewis, Victor Garcia Satorras, Bastiaan S Veeling, Frank Noé, et al. Improved motif-scaffolding with se (3) flow matching. ArXiv, 2024.
- Zhang & Skolnick (2004) Yang Zhang and Jeffrey Skolnick. Scoring function for automated assessment of protein structure template quality. Proteins: Structure, Function, and Bioinformatics, 57(4):702–710, 2004.
- Zhang & Skolnick (2005) Yang Zhang and Jeffrey Skolnick. Tm-align: a protein structure alignment algorithm based on the tm-score. Nucleic acids research, 33(7):2302–2309, 2005.
- Zheng et al. (2024) Shuxin Zheng, Jiyan He, Chang Liu, Yu Shi, Ziheng Lu, Weitao Feng, Fusong Ju, Jiaxi Wang, Jianwei Zhu, Yaosen Min, et al. Predicting equilibrium distributions for molecular systems with deep learning. Nature Machine Intelligence, pp. 1–10, 2024.
- Zheng et al. (2023) Zaixiang Zheng, Yifan Deng, Dongyu Xue, Yi Zhou, Fei Ye, and Quanquan Gu. Structure-informed language models are protein designers. In International conference on machine learning, pp. 42317–42338. PMLR, 2023.
- Zhou et al. (2024) Xiangxin Zhou, Dongyu Xue, Ruizhe Chen, Zaixiang Zheng, Liang Wang, and Quanquan Gu. Antigen-specific antibody design via direct energy-based preference optimization. In ICML 2024 Workshop AI4Science, 2024.